Publié le 17 mai 2007Lecture 15 min
Les formes frontières de la sclérose en plaques
Jérôme de SEZE, Clinique Neurologique, Hôpital Civil, Strasbourg
Plus d’un siècle après les premières descriptions de la sclérose en plaques (SEP), la cause exacte de la maladie reste inconnue. Ceci explique les descriptions de formes cliniques ou neuropathologiques particulières, appelées « formes frontières de SEP ». Dans cet article, nous décrirons successivement les formes pseudo-tumorales (dites de Balo, de Marburg ou de Schilder), les formes débutant chez le sujet âgé de plus de 50 ans, et les affections apparentées (neuromyélite optique de Devic et encéphalomyélite aiguë disséminée).
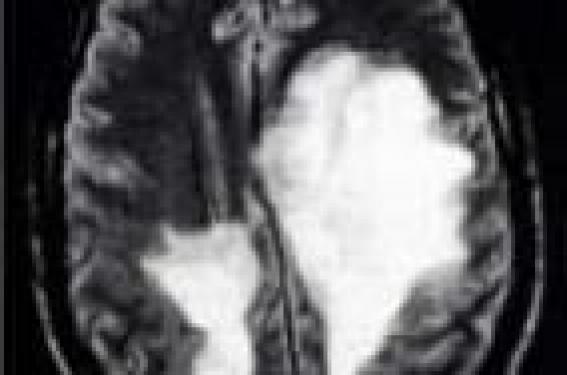
Malgré les progrès importants concernant la physiopathologie de la sclérose en plaques (SEP) avec l’avènement de l’IRM et des modèles animaux, un certain nombre d’interrogations persistent, notamment en ce qui concerne les formes frontières de la maladie. Faut-il considérer les formes pseudo-tumorales, les formes de l’enfant ou le syndrome de Devic comme des SEP atypiques ou comme des entités séparées ? Ce débat a-t-il une importance pour la prise en charge thérapeutique ou s’agit-il seulement d’une discussion nosologique ? Dans cette revue, nous traiterons successivement des formes pseudo-tumorales de SEP avec leurs différentes variantes anatomopathologiques et des formes cliniques apparentées comme le syndrome de Devic et l’encéphalomyélite aiguë disséminée (EMAD). Les formes pseudo-tumorales de SEP Les formes pseudo-tumorales de SEP sont rares et de diagnostic difficile, notamment lorsqu’il s’agit de l’épisode inaugural et qu’il n’existe qu’une lésion à l’IRM. Dans de tels cas, le diagnostic est fréquemment posé après une biopsie en condition stéréotaxique, réalisée dans le but d’éliminer une pathologie tumorale (gliome, lymphome, etc.). Le tableau clinique inaugural est souvent pseudo-vasculaire avec un déficit moteur sévère, parfois associé à une aphasie d’apparition brutale ou plutôt subaiguë. Le diagnostic est donc parfois discuté et retardé dans certains cas jusqu’à la biopsie. Le tableau clinique inaugural est souvent pseudo-vasculaire avec un déficit moteur sévère. Au sein des SEP pseudo-tumorales, il est classique de séparer trois cadres anatomopathologiques différents – la forme de Marburg, la forme de Schilder et la sclérose concentrique de Balo –, même si cette différenciation n’a le plus souvent pas de conséquence directe sur la prise en charge thérapeutique. La forme de Marburg est le plus souvent une atteinte démyélinisante suraiguë pouvant être monophasique. La forme de Schilder est essentiellement décrite en pédiatrie, mais il existe une certaine ambiguïté. En effet, elle ne correspond pas à une entité bien délimitée, et un certain nombre d’auteurs ont inclus sous ce terme des cas de leucodystrophies et d’autres affections inflammatoires juvéniles initialement pseudo-tumorales, évoluant finalement vers une SEP (P. Schilder, 1912). En cas de biopsie cérébrale, l’anatomopathologiste devrait donc être orienté non seulement vers une SEP, mais également vers le diagnostic de leucodystrophie. Enfin, la forme de Balo comporte des caractéristiques anatomopathologiques propres faites d’alternance de zones de myéline saine et de zones de démyélinisation. Elle évolue fréquemment vers une SEP. Forme de Marburg La première description a été rapportée par Marburg en 1906 à propos de trois patients atteints d’encéphalopathie aiguë, pour lesquels les données anatomopathologiques montraient des lésions de démyélinisation identique à celles observées dans la SEP (O. Marburg, 1906). Les lésions observées regroupaient des éléments du même âge associant une infiltration cellulaire importante, un œdème, des astrocytes géants, mais pas de gliose astrocytaire. La parenté avec la SEP fut établie grâce à l’observation d’une démyélinisation respectant en grande partie les axones. L’évolution de cette affection est souvent monophasique avec, dans certains cas, une aggravation progressive sur quelques jours entraînant le décès (F. Giubelei et coll., 1997). L’IRM montre une ou plusieurs volumineuses lésions, rehaussées inconstamment et partiellement par l’injection de gadolinium (figure 1). Figure 1. IRM cérébrale d’un patient atteint d’une SEP aiguë de type Marburg. L’hypothèse de modifications post-traductionnelles de la protéine basique de la myéline (MBP), entraînant une variation de sa charge globale, a été avancée comme le principal mécanisme responsable de cette forme; mais ceci n’a été décrit qu’à partir d’un cas (DD. Wood et coll., 1996). Compte tenu du faible nombre de patients rapportés, il n’y a pas de données sur la thérapeutique, mais certains proposent de fortes doses de corticoïdes, éventuellement associées à un immunosuppresseur. Cependant, le caractère fréquemment monophasique de cette forme de SEP rend l’analyse de l’efficacité d’une thérapeutique extrêmement difficile. Forme de Schilder Décrite en 1912 chez une jeune fille, âgée de 14 ans, décédée dans un tableau d’hypertension intracrânienne (Schilder, 1912), cette forme ne représente pas une entité neuropathologique particulière et le terme devrait être abandonné au profit de celui de SEP pseudo-tumorale, plus simple et plus général. En effet, l’analyse de la littérature retrouve sous ce terme des tableaux totalement différents dans leur présentation clinique et anatomopathologique, allant de la SEP pseudo-tumorale jusqu’aux leucodystrophies (P. Schilder, 1912 ; MJ. Fitzgerald et LT. Coleman, 2000). Ce terme est actuellement essentiellement utilisé par les neuropédiatres (ML. Fitzgerald et LT. Coleman, 2000) pour qualifier les SEP avec des lésions extensives pouvant couvrir pratiquement l’ensemble d’un hémisphère, voire envahir l’hémisphère controlatéral par l’intermédiaire du corps calleux (figure 2). Figure 2. IRM cérébrale d’une jeune fille atteinte d’une SEP diffuse décrite par P. Schilder. L’évolution se fait habituellement vers une SEP plus classique et de nouvelles poussées sur le même mode sont exceptionnellement observées. Un traitement par fortes doses de corticoïdes (5 à 10 g) est généralement proposé. En revanche, le traitement à moyen et long terme n’est pas codifié, ces formes n’ayant pas nécessairement un mauvais pronostic. Il semble donc logique d’instituer le même type de prise en charge que pour les SEP classiques en proposant un traitement de fond en cas de nouvel épisode neurologique ou de nouvelles lésions actives en IRM. Sclérose concentrique de Balo Décrite par J. Balo en 1928, chez un jeune patient atteint d’une hémiplégie et de troubles du langage, cette forme est particulière par son aspect de démyélinisation « en spirale » observé en neuropathologie et sur les IRM (figure 3). Figure 3 A, B et C. IRM cérébrale d’un patient atteint de sclérose concentrique de Balo montrant une lésion « striée » sous forme d’hypersignal en pondération T2 (A) rehaussé par le gadolinium (B et C). Ces spirales correspondent à une alternance de bandes de myéline normale et de zones démyélinisées. Ce type de lésions n’avait pas été jusqu’alors spécifiquement décrit dans les séries anatomopathologiques de la SEP de l’adulte récemment publiées (CF. Lucchinetti et coll., 2000). Un travail récent (C. Stadelmann et coll., 2005) a analysé les données autopsiques de 14 patients avec des lésions de type « Balo ». Les auteurs ont mis en évidence, à proximité des lésions actives et à moindre degré au niveau des jonctions entre myéline saine et pathologique, des protéines impliquées dans le préconditionnement telles que les protéines de haut choc thermique (HSP-70) ou des protéines inductrices d’hypoxie (hypoxia-inducible factor 1 a). Cependant, si la sclérose concentrique de Balo a tout d’abord été considérée comme une entité autonome, différentes études ont mis en évidence la coexistence de lésions de type « Balo » à des lésions classiques de SEP (Y. Kuroiwa, 1985 ; C. Stadelmann et coll., 2005). L’évolution peut être sévère, comme pour le cas princeps de Balo, décédé en 4 mois, mais peut également être plus favorable et se faire vers une SEP plus habituelle. Compte tenu du faible nombre de patients rapportés dans la littérature, aucune attitude thérapeutique n’a démontré une efficacité supérieure à une autre. Mais, certains auteurs préconisent la mise en route rapide de fortes doses de corticoïdes pouvant aller jusqu’à 10 g de méthylprednisolone intraveineux. D’autres proposent l’utilisation d’immunosuppresseurs d’emblée afin de diminuer les risques de séquelles, fréquentes dans ce tableau clinique. L’ensemble de ces trois formes particulières de SEP pseudo-tumorales ne se différencie finalement de la SEP classique que par les données anatomopathologiques et IRM. L’usage de noms propres pour séparer ces formes cliniques ne semble pas à l’heure actuelle d’un grand intérêt, tant sur le plan diagnostique que thérapeutique et pourrait être remplacé par le terme de SEP pseudo-tumorale, plus simple et plus explicite. Cependant, des incertitudes persistent à la fois sur le plan thérapeutique et sur l’évolution qui peut être soit monophasique, soit par poussées. Une étude multicentrique, regroupant ces cas, pourrait tenter de répondre aux questions suivantes : faut-il traiter ces patients par de fortes doses de corticoïdes à la phase aiguë de la maladie ou peut-on se contenter des doses habituelles de 3 à 5 g ? Faut-il proposer un relais par immunosuppresseur d’emblée ou peut-on attendre une hypothétique deuxième poussée pour juger de l’opportunité d’un traitement de fond par immunomodulateur ou immunosuppresseur en fonction de l’intensité clinique et/ou radiologique de la maladie ? L’ensemble de ces trois formes particulières de SEP pseudo-tumorale ne se différencie de la SEP classique que par les données anatomopathologiques et l’IRM. Formes cliniques apparentées (syndrome de Devic et EMAD) Le syndrome de Devic et l’EMAD représentent les deux entités dont l’apparenté à la SEP a été le plus discutée. Il s’agit de formes rares, mais non exceptionnelles : les données cliniques, radiologiques et anatomopathologiques de la littérature ont tenté de répondre à cette question. De toute évidence, il existe des sujets répondant à la fois aux critères de SEP et à ceux du syndrome de Devic ou d’EMAD. Ce débat n’aurait pas grande importance s’il n’en découlait des sanctions thérapeutiques éventuellement ciblées tant à la phase aiguë que par la suite. Maladie de Devic Dès 1894, E. Devic décrivait un tableau clinique très particulier touchant la moelle et les nerfs optiques. Plus d’un siècle après les premières descriptions, un grand nombre de questions concernant ce syndrome subsistent : doit-on considérer cette affection comme un syndrome ou une maladie ? Faut-il traiter le syndrome de Devic comme une SEP ? Il existe des données cliniques, paracliniques et anatomopathologiques propres au syndrome de Devic. Sur le plan clinique, l’atteinte médullaire est sévère, de type myélite transverse, associée à une atteinte ophtalmologique uni- ou bilatérale, pouvant dans les cas extrêmes conduire à un tableau de tétraplégie et de cécité. Sur le plan paraclinique, on note habituellement une lésion étendue à plus de trois métamères vertébraux (figure 4). Figure 4 A et B. IRM médullaire chez une patiente atteinte d’une neuromyélite optique de Devic montrant un hypersignal étendu de plus de 3 segments vertébraux avec hypertrophie médullaire (A : coupe sagittale), touchant la totalité de la moelle à point de départ centro-médullaire (B : coupe axiale). La ponction lombaire montre le plus souvent une hypercytose supérieure à 50 éléments associée à une hyperprotéinorrachie supérieure à 1 g, sans profil oligoclonal à l’immuno-électrophorèse. Des critères diagnostiques du syndrome de Devic ont été proposés, incluant ces différents paramètres cliniques et paracliniques (DM. Wingerchuk et coll., 1999). Ces critères permettaient dans la plupart des cas de différencier la SEP et le syndrome de Devic (J. de Seze et coll., 2003). La découverte récente d’un anticorps (anticorps anti-NMO), retrouvé chez plus de 70 % des patients atteints de syndrome de Devic et chez moins de 10 % de patients atteints de SEP (VA. Lennon et coll., 2004) a amené cette équipe (DM. Wingerchuck et coll., 2006) à proposer de nouveaux critères simplifiés en s’appuyant sur les données immunologiques et IRM (tableau). Ces anticorps anti-NMO sont dirigés contre l’aquaporine 4 (AQP4), protéines des canaux hydriques, ubiquitaires, mais dont la concentration est importante dans la moelle et le nerf optique (VA. Lennon et coll., 2005). Les données IRM bien corrélées avec les résultats neuropathologiques d’une étude récente de C. Lucchinetti et coll. (2002) montrent des lésions étendues, nécrotiques, associées à des dépôts de lymphocytes B. Ceci, associé à la découverte des anticorps anti-NMO, confirme la prédominance de l’atteinte humorale sur l’atteinte cellulaire dans le syndrome de Devic, déjà évoquée par son association fréquente à certaines maladies dysimmunitaires ou endocriniennes (JI. O’Riordan et coll., 1996 ; JC. Vernant et coll., 1997 ; DM. Wingerchuk et coll., 1999 ; J. de Seze et coll., 2002). Toutes ces distinctions entre la SEP et la neuromyélite optique de Devic n’auraient qu’un intérêt nosologique s’il n’en découlait des traitements potentiellement différents. Devant la plus grande sévérité du syndrome de Devic, un certain nombre d’auteurs proposent d’utiliser d’emblée et systématiquement un traitement immunosuppresseur de type cyclophosphamide ou mitoxantrone (de J. Seze et coll., 2003 ; P. Cabre et coll., 2003). En effet, une étude rétrospective récente montre que le pronostic à long terme est plus péjoratif chez les sujets qui n’ont pas reçu d’immunosuppresseurs comparés à ceux traités par cyclophosphamide ou mitoxantrone (P. Cabre et coll., 2003). Enfin, un travail récent rapporte l’efficacité du rituximab dans le syndrome de Devic (BA. Cree et coll., 2005). Cependant, ces résultats méritent d’être confirmés par des études contrôlées. Un travail récent rapporte l’efficacité du rituximab dans le syndrome de Devic. EMAD Cette affection touche essentiellement les sujets jeunes, le plus souvent dans un contexte post-infectieux. Le délai entre l’infection et le phénomène réactionnel inflammatoire est habituellement de 1 à 4 semaines. Les signes cliniques s’installent de façon rapidement progressive, incluant des céphalées et un syndrome fébrile, des signes neurologiques focaux avec une atteinte médullaire fréquente d’où le terme d’encéphalomyélite. Il n’y a cependant pas de critère diagnostique d’EMAD, rendant difficile sa description en une seule entité clinique. En effet, certains auteurs utilisent également ce terme pour qualifier des tableaux de vasculite post-infectieuse. Il n’y a pas de critère diagnostique d’EMAD, rendant difficile sa description en une seule entité clinique. Une méningite lymphocytaire avec une discrète hyperprotéinorrachie est habituellement associée au tableau clinique, le plus souvent sans profil oligoclonal à l’électrophorèse des protéines. L’IRM cérébrale montre des lésions hypersignal en pondération T2, parfois hyposignal en pondération T1, de même âge, souvent rehaussées par le produit de contraste (figure 5). Figure 5 A et B. IRM cérébrale d’une patiente atteinte d’une encéphalomyélite aiguë disséminée montrant des hypersignaux multiples en séquence pondérée T2, d’âge identique (A), prenant pour la plupart d’entre eux le produit de contraste (B). L’évolution est habituellement monophasique, mais dans certains cas, des rechutes peuvent être observées dans les six mois suivant les symptômes initiaux et ne doivent pas remettre en cause le diagnostic (Mansini et coll., 1996). Par ailleurs, quelques cas ont évolué secondairement vers une SEP, notamment chez les patients ayant un profil oligoclonal à la ponction lombaire (RC. Dale et coll., 2000 ; S. Schwarz et coll., 2001 ; JL. Hynson et coll., 2001). Cette distinction est actuellement très importante compte tenu des possibilités thérapeutiques, notamment à la phase précoce des maladies inflammatoires. En effet, dans les cas d’EMAD typiques contrairement à la SEP, le traitement n’est pas codifié. Il repose généralement sur de fortes doses de corticoïdes, parfois associées à des plasmaphérèses (R. Miyazawa et coll., 2001). Par analogie avec d’autres pathologies post-infectieuses ou post-vaccinales, un traitement par immunoglobulines a également été proposé sans preuve évidente de son efficacité (E. Marchioni et coll., 2002). L’évolution habituelle conduit à une récupération complète des symptômes et à une absence de récidive. Même si un grand nombre de données cliniques et paracliniques sont proches entre ces différentes formes frontières de SEP, ces distinctions nosologiques ont une portée anatomopathologique et physiopathologique qui pourrait avoir, dans les années à venir, une importance dans les décisions thérapeutiques, à condition de pouvoir regrouper suffisamment de patients pour des études thérapeutiques. Conclusion Il semble qu’un certain nombre d’entités précédemment évoquées devraient être distinguées de la SEP (syndrome de Devic, EMAD monophasique) et d’autres intégrées dans le groupe des SEP sans distinction particulière notamment dans l’approche thérapeutique (EMAD évoluant vers une SEP). D’une manière générale, la SEP reste probablement composée de plusieurs entités, et la discussion nosologique n’en est que plus intéressante dès lors qu’elle permet de mieux identifier les sous-types de SEP. Il subsiste cependant un certain nombre de questions devant de tels tableaux cliniques notamment sur le plan pronostique et thérapeutique : lors de l’épisode initial, quel est le risque de récidive et/ou d’évolution vers une SEP ? Quelle est la thérapeutique adaptée à ces patients tant à la période aiguë (très fortes doses de corticoïdes, plasmaphérèse, immunoglobulines, immunosuppresseurs d’emblée) qu’à la phase chronique (abstention, immunomodulateur, immunosuppresseur) ? Nous remercions de S. Wiertlewski (CHU de Nantes) qui a fourni l’IRM de la figure 3.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
