Publié le 20 sep 2006Lecture 17 min
Origines de la SEP : apports de l'épidémiologie
Isabelle Cournu-Rebeix, INSERM UMR 546, Faculté de médecine pitié salpêtriere (Paris)
Décrite depuis le milieu du XIX e siecle, la sclérose en plaques (SEP) est une affection chronique, inflammatoire et démyelinisante du systéme nerveux central dont la cause demeure inconnue. Il est de nos jour admis qu'une interaction gène-environnement est à l'origine du developpement de la maladie. Des etudes épidemiologiques et génétiques ont permis de révéler que le processus immunitaire impliqué dans la sléroses en plaques ne se déclenche que chez les sujets génétiquement prédisposés après exposition à un ou plusieurs facteurs environnementaux.
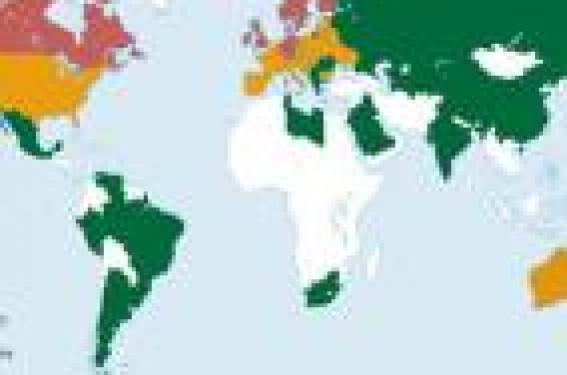
Physiopathologie et symptomatologie Les plaques, qui ont donné leur nom à la maladie sont des lésions focales du système neveux central (SNC) qui prédominent dans la substance blanche. Elles se caractérisent par un infiltrat de cellules immunitaires, témoin d'un processus inflammatoire et par une perte totale ou partielle de myéline. Au sein de ces lésions, on observe des atteintes axonales dont on ne connaît pas le potentiel de réversibilité mais dont la chronicité pourrait conduire au handicap neurologique. Des processus de réparation des lésions myéliniques (remyélinisation) peuvent être observés, mais ils restent le plus souvent partiels. La symptomatologie révélatrice de la sclérose en plaques est très variée et dépend de l'âge du patient. Les symptômes visuels et sensitifs prédominent chez le sujet jeune, tandis que l'atteinte motrice est plus fréquente chez le sujet âgé. Le diagnostic de SEP est fondé sur la mise en évidence clinique et/ou paraclinique d'au moins deux lésions de démyélinisation, affectant différents sites du cerveau ou de la moelle épinière et disséminées dans le temps. Afin de déterminer la répartition des lésions dans le temps et dans l'espace, plusieurs outils diagnostiques ont été validés, pour appuyer l'interrogatoire et l'examen clinique : l'analyse du liquide céphalo-rachidien, la mesure des potentiels évoqués et l'IRM. Dans l'effort mené pour com-prendre l'étiologie de la SEP, il a été fortement suggéré que les lésions tissulaires observés dans cette maladie et les symptômes cliniques seraient dus à une attaque autoimmune de la myéline et des cellules myélinisantes (oligodendrocytes) du SNC. Cependant, le caractère autoimmun de la SEP reste encore controversé, faute d'un auto-antigène cible clairement identifié. SEP : une maladie multifactorielle La répartition de la sclérose en plaques à travers le monde n'est pas uniforme. La prévalence (nombre de cas connus à un moment donné au sein d'une population) dépend de la latitude et croît, dans chaque hémisphère, lorsque l'on s'éloigne de l'équateur. L'interprétation de l'existence de ce gradient nord-sud de la sclérose en plaques, confirmée par de nombreuses études épidemiologiques n'est pas univoque (figure 1). Figure 1. Répartition mondiale de la SEP (prévalence pour 100 000 habitants). (D'après Ruth AM. Lancet Neurol 2004). Un ou plusieurs facteurs environnementaux prédominant aux antipodes pourraient expliquer cette répartition dans les zones de populations homogènes telles que l'Australie et la Nouvelle-Zélande. Toutefois, cette hypothèse environnementale n'est pas suffisante pour rendre compte des fortes variations de prévalence observées au sein d'un même pays ou à une même latitude (tableau 1). Les fortes différences de prévalence observées au sein d'une même zone géographique, en fonction de l'origine ethnique, plaident en faveur de l'existence de prédispositions génétiques différentes. Épidémiologie environnementale Les facteurs environnementaux Les hypothèses concernant la nature de la composante environnementale de la SEP sont nombreuses et variées. Des facteurs comme le climat (ensoleillement, température, degré d'humidité), la teneur en métaux lourds de l'eau et du sol, la pollution, ont été incriminés à partir de corrélations géographiques avec la prévalence (nombre de cas recensés à un moment donné pour 100 000 habitants)1. Toutefois même si des mécanismes étiopathogéniques ont pu être proposés pour expliquer certaines corrélations, telle qu'une action des métaux lourds sur le système immunitaire, ou l'influence du degré d'exposition solaire sur la production de vitamine D, aucune de ces études n'a permis ni de valider ni d'exclure l'un ou l'autre de ces facteurs comme facteur de risque dans la SEP. Certaines études cas-témoins ont rapporté une association entre la sclérose en plaques et des facteurs environnementaux. Ainsi le risque de développer la maladie a été alternativement associé à la culture de l'avoine, à l'exploitation de troupeaux de bétail, à l'emploi des solvants organiques, à l'industrie du papier, au travail du cuir ou encore aux métiers de la laine. Toutefois, ces études ne portaient que sur de très petits échantillons et n'ont pour la plupart jamais été répliquées. Les facteurs infectieux L'hypothèse selon laquelle un agent infectieux jouerait un rôle dans la survenue de la sclérose en plaques suscite beaucoup d'intérêt depuis plus d'un siècle. En 1884, Pierre Marie parlait déjà d'une coïncidence d'occurrence entre la SEP et les maladies infectieuses. Cependant il est encore impossible aujourd'hui d'affirmer ou d'infirmer une participation infectieuse à l'étiopathologie de la SEP. Plusieurs agents infectieux, en particulier des virus, sont capables d'induire une réponse auto-immune similaire à celle suspectée dans la SEP. Les principaux mécanismes de cette induction seraient le mimétisme moléculaire (homologie de séquences entre un antigène exogène et un autoantigène) ou la superantigénicité (antigène capable d'activer tous les lymphocytes T d'un hôte sans spécificité). Il est encore impossible aujourd'hui d'affirmer ou d'infirmer une participation infectieuse à l'étiopathologie de la SEP. Au plan purement épidémiologique, les résultats des études de migrants supportent aussi une implication virale dans la SEP. Des études de prévalence ont été réalisées sur des groupes de Nord-Européens et de Nord-Américains migrant de leur pays d'origine (zones à haute prévalence) vers des zones de moindre risque comme Israël ou l'Afrique du sud. Ces études ont montré que les individus migrant après l'âge de 15 ans conservaient un risque élevé de développer la maladie, similaire à celui observé dans leur pays d'origine. En revanche, la prévalence était similaire à celle du pays d'accueil pour les enfants migrant avant l'adolescence. La différence de risque corrélée à l'âge de migration semble indiquer qu'il existe un âge critique d'exposition aux facteurs environnementaux qui s'accompagne d'un délai important entre l'acquisition et l'apparition clinique de la maladie. Cette latence pourrait s'expliquer par une ré-exposition à des infections virales particulières contractées au cours de l'enfance. Même si ces études sont affaiblies par le petit nombre d'individus explorés et l'hétérogénéité potentielle entre les migrants et leur population d'origine, elles constituent par leur nombre et la concordance de leurs résultats, un argument solide en faveur d'un agent causal infectieux. Toutefois, les études cas-témoins déjà menées n'ont pas permis de détecter de différences dans le nombre ou le type de maladies virales contractées au cours de l'enfance entre les patients atteints de SEP et les témoins. En revanche, les patients semblent développer des maladies virales infantiles à un âge plus tardif que les individus sains. Cette constatation peut expliquer une plus faible incidence (nombre de nouveaux cas apparaissant au sein d'une population pendant une durée donnée) de la SEP chez les individus issus de pays sous-développés ou de milieux socio-économiques défavorisés. En effet, les conditions de vie défavorables et l'insuffisance des infrastructures sanitaires favoriseraient l'éclosion plus précoce de maladies virales infantiles. Une grande proportion d'enfants habitant des zones de faible prévalence possède un titre d'anticorps élevé contre plusieurs maladies virales très précocement. L'hypothèse la plus probable découlant de ces observations est que la SEP serait une séquelle retardée à une ou plusieurs expositions virales infantiles. Une infection à un âge précoce pourrait être protectrice, alors qu'une infection chez un individu au système immunitaire mature augmenterait le risque de développer une SEP. Plusieurs virus humains et animaux ont été suspectés d'être des facteurs déclenchants de la sclérose en plaques. La présence de taux élevés d'anticorps antiviraux dans le sérum et le LCR des patients a permis d'incriminer les virus suivants : le virus de Theiler, le virus de la rougeole, celui de la roséole (herpes-virus humain 6), des oreillons, de la varicelle, l'Epstein-Barr virus ou encore le HTLV-1 (virus responsable des paraparésies tropicales spastiques). Cependant, aucune de ces études de corrélation n'a pu démontrer de façon certaine le rôle causal d'un virus particulier dans le développement de la SEP. En revanche, il a été montré que les infections virales jouaient un rôle dans la progression neurologique de la maladie. Selon une étude publiée en 1985, le taux annuel de poussées chez les patients est trois fois plus élevé pendant les périodes d'infections virales. Une corrélation entre les infections virales respiratoires et l'apparition de poussées a également été démontrée. Certains auteurs ont émis l'hypothèse selon laquelle l'histoire naturelle de la sclérose en plaques (poussées/remissions) pouvait s'expliquer par une infection virale persistante de certains virus tel que l'Epstein Barr qui se caractérise par l'alternance de périodes de latence et de réactivation. La SEP serait-elle une séquelle retardée à une ou plusieurs expositions virales infantiles ? Épidémiologie génétique Les formes familiales de SEP sont rares : seulement 10 à 15 % des patients ont un apparenté atteint. Les études épidémiologiques classiques se basent sur des apparentés spécifiques des patients, tels que les descendants directs, les ascendants directs, les frères et sœurs et en particulier les jumeaux (monozygotes ou dizygotes). Ces études ont pour but de déterminer le taux de concordance pour la maladie chez les apparentés et de le comparer à celui de la population générale2 (tableau 2). Le risque de développer une SEP est corrélé au pourcentage de matériel génétique partagé, ce qui plaide en faveur de l'existence d'une prédisposition génétique à la sclérose en plaques. Le but des études génétiques de la SEP est de déterminer quels sont les facteurs génétiques impliqués dans le développement mais aussi dans la sévérité de la maladie. En effet, des études ont montré une corrélation significative de l'indice de progression de la maladie entre germains atteints, suggérant que des facteurs familiaux et peut-être génétiques interviennent dans la sévérité de la SEP3. Le risque de développer une SEP est corrélé au pourcentage de matériel génétique partagé avec un sujet apparenté atteint. L'identification et la localisation de facteurs de risque génétiques nécessitent l'utilisation de marqueurs génétiques (sites naturellement polymorphes de l'ADN). Si les malades d'une même famille partagent un allèle particulier d'un marqueur plus souvent que ne le voudrait le hasard, il existe une liaison génétique entre le locus étudié et la maladie (étude de familles multi-cas). Si des malades non apparentés sont plus souvent porteurs d'un allèle particulier d'un marqueur donné que des individus sains, il existe alors une association entre le marqueur étudié et la maladie (étude de familles mono-cas). Utilisant ces méthodes statistiques (liaison et/ou association), deux approches sont classiquement mises en œuvre dans la recherche de facteurs de prédisposition à la SEP : – les criblages anonymes du génome (figure 2) ; – l'approche gène candidat (figure 3). Figure 2. Recherche de facteurs de prédisposition génétique de la SEP par criblage anonyme du génome. Figure 3. Recherche de facteurs de prédisposition génétique par étude d’un gène candidat. L'association SEP/Complexe majeur d'histocompatibilité (CMH) L'association entre la sclérose en plaques et la région du CMH (ou HLA) fut décrite pour la première fois en 1972 sur la base d'études sérologiques et cellulaires4. De nombreux travaux fondés sur des analyses sérologiques (anticorps anti-HLA), ou plus récemment sur des analyses moléculaires, ont confirmé et précisé cette association. La spécificité HLA-DR2 est fortement associée à la SEP dans les populations caucasiennes d'Europe du Nord, alors que dans les populations sarde, jordanienne et japonaise, l'association est retrouvée avec la spécificité HLA-DR4. Bien qu'une association entre l'haplotype HLA-DRB1*1501-DQA1*0102-DQB1*0602 ait été montrée, la localisation précise de l'allèle responsable de la prédisposition au sein de la région DR, DQ n'a pas encore abouti. D'un point de vue fonctionnel, certains allèles des antigènes HLA pourraient conduire à une incapacité du CMH à sélectionner négativement les cellules T autoréactives ou à conférer une plus grande affinité aux lymphocytes T autoréactifs pour les antigènes myéliniques. Les autres gènes de prédisposition à la sclérose en plaques L'association et la liaison entre le locus HLA et la maladie ne peuvent expliquer à elles seules la corrélation familiale observée. Il semble que ce locus ne soit responsable qu'au maximum de 15 % de l'accroissement du risque de SEP pour les germains d'individus atteints. La prédisposition génétique à la SEP apparaît déterminée par plusieurs gènes indépendants ou interactifs, exerçant chacun un effet modeste5. Des analyses de ségrégation, réalisées dans des familles comportant plusieurs membres atteints n'ont pas permis d'identifier de gène majeur de prédisposition à la SEP. On ignore à l'heure actuelle, le nombre de gènes impliqués dans le processus pathologique et leur mode de transmission. L'association et la liaison entre le locus HLA et la maladie ne peuvent expliquer à elles seules la corrélation familiale observée. Sept études de criblages anonyme du génome, fondées sur des analyses de liaison ont été publiées de 1996 à 2002. Bien que chacun de ces criblages ait suggéré de nombreuses régions chromosomiques potentiellement liées à la SEP, peu d'entre elles sont communes à plusieurs études. D'autre part, des études de réplication ou des méta-analyses n'ont pas permis dans la plupart des cas d'aboutir aux mêmes conclusions. Le manque de résultats significatifs et communs de ces études par criblage systématique du génome dans la sclérose en plaques, renforce l'idée selon laquelle, il n'existe pas de locus à effet majeur responsable du développement de cette maladie. Au cours de ces dernières années de nombreuses études gènes-candidats ont été menées dans la SEP. Les gènes ont pour la plupart été choisis par référence à l'aspect neuropathologique des plaques qui associe une réaction inflammatoire et une démyélinisation, et à, l'existence d'un modèle animal : l'encéphalomyélite allergique expérimentale (EAE). Bien que la liste des gènes candidats testés soit longue, il n'existe pas à ce jour, de facteurs génétiques, autre que HLA, dont le rôle soit établi avec certitude dans la SEP. Nous n'aborderons que quelques exemples. Les gènes codant pour les protéines de la myéline font l'objet d'études depuis de nombreuses années. Des études de réplication portant sur de nouveaux polymorphismes et un plus grand nombre de familles n'ont pas permis de les impliquer de façon certaine dans le développement de la SEP. Si l'on prend l'exemple de la « Myelin Basic Protein » (MBP), sur quatorze études menées entre 1990 et 2003, six ont conclu à une association et/ou une liaison entre la MBP et la SEP, huit ont abouti à des résultats négatifs. Les gènes codant pour des protéines impliquées dans la réaction immunitaire ont été testés avec des résultats contradictoires. Une mutation ponctuelle interférant avec la maturation de l'ARNm a été mise en évidence dans le gène codant pour une tyrosine phosphatase (CD45). Cette mutation pourrait être associée à la SEP dans un faible nombre de cas (moins de 10 %). Concernant les cytokines et leurs récepteurs, un variant du récepteur à l'IL4 pourrait être associé aux formes primaires progressives de la maladie. Le rôle du gène codant pour la chaîne bêta du récepteur à l'IL2, pourrait être associé à la SEP, une étude de réplication est en cours. La molécule CTLA-4 est impliquée dans les phases terminales de l'activation des lymphocytes T. Il semble exister une association entre certains variants du gène codant pour cette protéine et la prédisposition génétique à la SEP. n Les molécules d'adhésion sont indispensables au passage des lymphocytes à travers la barrière hémato-encéphalique, évènement précoce dans le développement de la SEP. Un haplotype particulier des gènes codant pour l'Intercellular Adhesion Molecule 1 ( ICAM-1) pourrait jouer un rôle protecteur contre la maladie, ces résultats restent à confirmer. ApoE est un facteur génétique bien connu de prédisposition à la maladie d'Alzheimer. Certains auteurs se sont interrogés sur son implication dans la sévérité de la SEP. Les conclusions de ces études sont cependant contradictoires. Les facteurs de croissance des oligodendrocytes, la cellule myélinisante du système nerveux central, ont été testés comme gènes de prédisposition à la SEP. Seul le TGFb3 pourrait contribuer à l'apparition de la maladie, une étude de réplication est en cours. Conclusion Les approches épidemiologiques de la sclérose en plaques, même si elles n'ont pas permis jusqu'à présent de mettre en évidence l'implication de facteurs spécifiques ont révélé des informations majeures. L'idée selon laquelle la SEP résulterait d'une interaction gène-environnement est aujourd'hui largement admise, de même que le caractère polygénique de la maladie. La composante environnementale semble agir au niveau d'une population plutôt que sur le micro-environnement familial alors qu'une plus forte concordance pour la maladie au sein d'une famille résulterait du partage de matériel génétique. Dans le domaine de la génétique, la seule certitude à l'heure actuelle, est l'existence d'un facteur de prédisposition à cette maladie dans la région du complexe majeur d'histocompatibilité sur le bras court du chromosome 6. Les nombreuses études menées depuis une trentaine d'année, criblages systématiques du génome ou études de gènes candidats, n'ont pas permis d'identifier d'autres facteurs de façon certaine. Peut-être est-il temps, d'envisager de nouvelles stratégies ? Le développement récent des puces à ADN a permis de dresser une liste de gènes dont l'expression est fortement augmentée ou diminuée au sein des lésions de SEP en comparaison aux zones cérébrales non lésées. Cette liste, bien que nécessitant une validation, constitue un réservoir intéressant de gènes candidats non plus seulement par fonction mais aussi par variation d'expression. Par ailleurs, la quantité importante de données épidemiologiques et génétiques engendrées par les nombreuses études menées permettent aujourd'hui de pouvoir tester le rôle, non plus d'un facteur isolé mais d'une combinaison de facteurs épidemiologiques et/ou génétiques.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :
