Publié le 26 jan 2009Lecture 8 min
Retard psychomoteur : quand évoquer une pathologie mitochondriale ?
M. RIO, P. DE LONLAY, A. MUNNICH, Hôpital Necker-Enfants malades Paris
Les maladies mitochondriales regroupent une grande variété de pathologies dont le dénominateur commun est l’existence d’un déficit de la chaîne respiratoire mitochondriale. Chez l’enfant, une hypotonie et/ou un retard psychomoteur peuvent être le symptôme révélateur de la maladie. Or, l’identification de la cause d’une hypotonie ou d’un retard psychomoteur est essentielle pour répondre aux demandes d’explication et de pronostic des parents, pour orienter la prise en charge mais aussi permettre un conseil génétique. Nous présentons ici les signes cliniques, métaboliques et radiologiques qui permettront d’orienter vers le diagnostic de déficit de la chaîne respiratoire mitochondriale.
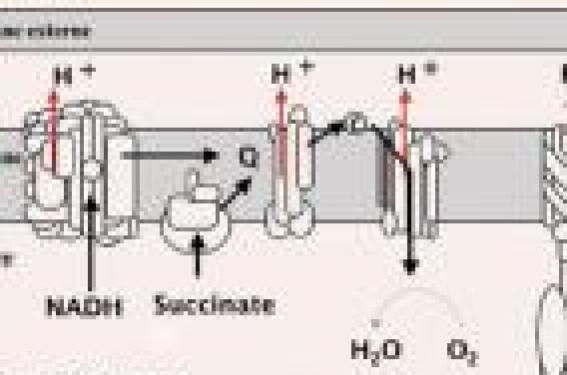
Le fonctionnement d’un grand nombre de tissus dépendant de la fourniture en énergie par la chaîne respiratoire mitochondriale, un déficit de cette dernière peut théoriquement donner lieu à n’importe quel symptôme dans n’importe quel organe ou tissu Physiologie La mitochondrie assure la respiration cellulaire et la production d’énergie grâce à la phosphorylation oxydative. Celle-ci a lieu au niveau de la chaîne respiratoire mitochondriale composée de cinq complexes multienzymatiques situés dans la membrane interne de la mitochondrie. Les quatre premiers complexes fonctionnent comme transporteurs d’électrons à partir de réactions d’oxydation de différents substrats et aboutissent à la consommation d’oxygène par le complexe IV. Le complexe V assure la synthèse d’adénosine-triphosphate (ATP) à partir de l’adénosine-diphosphate (ADP) et du phosphate inorganique dans la matrice mitochondriale par une réaction de phosphorylation (figure 1). La chaîne respiratoire a une double origine génétique : nucléaire et mitochondriale (figure 2). Figure 1. Chaîne respiratoire mitochondriale. NADH : nicotinamide adénosine dinucléotide ;ATP : acide adénosine-triphosphate ; ADP : acide adénosine-diphosphate ; Pi : phosphate inorganique ;CI, CII, CII, CIV, CV : complexes I, II, III, IV, V de la chaîne respiratoire mitochondriale ; Q : ubiquinone. Figure 2. La double origine génétique de la chaîne respiratoire mitochondriale. CI,CII, CII, CIV, CV : complexes I, II, III, IV,V de la chaîne respiratoire mitochondriale ; ADNmt : ADN mitochondrial. Présentation cliniques La chaîne respiratoire mitochondriale ayant pour rôle la synthèse d’ATP nécessaire à toutes les cellules de l’organisme, un déficit de la chaîne respiratoire peut donner théoriquement lieu à n’importe quel symptôme dans n’importe quel organe ou tissu. Ainsi, le diagnostic de maladie mitochondriale doit être évoqué chez des patients présentant une atteinte inexpliquée, neuromusculaire ou non, associant des organes apparemment sans relation, en dehors du fait que chacun d’entre eux possède des mitochondries. L’atteinte du système nerveux central peut être chez le nourrisson et l’enfant le mode d’entrée dans la maladie. La possibilité d’une cytopathie mitochondriale doit être envisagée lorsque s’associe à l’hypotonie ou au retard psychomoteur tout autre symptôme illégitime. Une liste non exhaustive des signes cliniques qui doivent faire évoquer le diagnostic de cytopathie mitochondriale lorsqu’ils s’associent à un retard psychomoteur est présentée au tableau 1. Tableau 1. Signes cliniques et biologiques devant faire évoquer une maladie mitochondriale. Coeur • Myocardiopathie • Troubles du rythme Oeil • Paralysie oculomotrice • Atrophie optique • Ptosis • Cataracte Système nerveux • Syndrome cérébelleux • Régression psychomotrice • Epilepsie • Pseudo-accident vasculaire • Migraine • Déficit musculaire • Myalgies Audition • Surdité Rein • Tubulopathie • Néphropathie tubulo-interstitielle Foie • Hépatomégalie • Cytolyse • Insuffisance hépatique Glandes endocrines • Diabète insulinodépendant • Diabète insipide • Déficit en hormone de croissance • Hypoparathyroïdie Appareil digestif • Cassure staturopondérale • Anorexie • Vomissements • Diarrhée Il existe des associations cliniques plus fréquentes et qui ont été identifiées en tant qu’entités distinctes. Certaines d’entre elles peuvent se rencontrer chez l’enfant ou l’adolescent et s’accompagner d’une atteinte du système nerveux central ; elles sont décrites dans le tableau 2. Tableau 2. Entités cliniques distinctes liées à un remaniement de l’ADNmt. Syndrome Phénotype clinique Syndrome NARP (Neurogenic Ataxia Retinitis ataxie cérébelleuse, Pigmentosa) Neuropathie sensorielle, ataxie cérébelleuse, rétinopathie pigmentaire Syndrome MELAS (Mitochondrial Encephalomyopathy-Lactic musculaire, pseudo-Acidosis-Stroke like episodes) Migraines récurrentes, vomissements, faiblesse accidents vasculaires cérébraux, acidose lactique, régression psychomotrice possible Syndrome MERFF (Myoclonus Epilepsy Ragged Red Fibers) Encéphalomyopathie avec myoclonies, ataxie, surdité et faiblesse musculaire, régression Syndrome Kearns-Sayre Ophtalmoplégie externe progressive, rétinite pigmentaire, régression psychomotrice, trouble de la conduction cardiaque Il est à noter que tous ces syndromes sont liés à un réarrangement de l’ADN mitochondrial (ADNmt). L’association d’un symptôme « illégitime » à une hypotomie ou à un retard mental doit faire envisager une cytophatie mitochondriale. Examens complémentaires d’orientation Lorsqu’une cytopathie mitochondriale est suspectée, un bilan d’investigation doit être réalisé (encadré). Il comporte l’exploration systématique des autres organes à la recherche d’une atteinte multiviscérale et un bilan métabolique. Bilan d’investigation d’une cytopathie mitochondriale Recherche d’atteintes viscérales multiples • Échographie cardiaque • Examen ophtalmologique avec fond d’oeil, électrorétinogramme et potentiels évoqués visuels • Audiométrie • Bilan hépatique • Bilan de la fonction rénale • IRM cérébrale avec spectroscopie • IRM cérébrale avec spectroscopie Examens métaboliques d’orientation • Cycle Redox avec détermination des taux de lactate (L), pyruvate (P), rapport L/P, ammoniémie, acides gras libres (AGL), 3-hydroxybutyrate (3 OHB) et acétoacétate (AcAc), rapport 3 OHB/AcAc, rapport AGL/corps cétoniques et glucose • Chromatographies des acides aminés plasmatiques • Chromatographies des acides organiques urinaires L’apport du bilan métabolique Les anomalies suivantes sont évocatrices d’une maladie mitochondriale : – une acidose lactique ; – une hyperlactacidémie avec rapports L/P augmentés, élévation du rapport 3 OHB/AcAc, élévation des corps cétoniques en postprandial ; – une hyperalaninémie et hyperprolinémie ; – une lactaturie. Ces perturbations biochimiques doivent conduire à l’étude de la chaîne respiratoire mitochondriale. Toutefois, leur absence n’élimine pas le diagnostic. L’apport de l’IRM cérébrale et de la spectroscopie Outre l’atteinte bilatérale des noyaux gris centraux, les anomalies de signal du tronc cérébral et les anomalies du cervelet (atrophie progressive et/ou hypersignaux des noyaux dentelés) doivent faire évoquer une maladie mitochondriale. Ces anomalies sont d’autant plus évocatrices que s’y associe la présence de lactate dans les zones atteintes. Diagnostic Étude enzymologique Le diagnostic de maladie mitochondriale repose principalement sur l’étude enzymatique des différents complexes de la chaîne respiratoire. Cette étude se fait par des techniques de polarographie et de spectrophotométrie. Elle doit être réalisée dans la mesure du possible sur le tissu atteint. Toutefois, lorsque des organes d’accès difficile sont touchés (cerveau, rétine, système endocrinien), les investigations ne pourront se faire que sur des tissus périphériques, le plus souvent le muscle squelettique. Quel que soit l’organe atteint, il est essentiel de pratiquer une biopsie de peau. En effet, de cette étude sur fibroblastes en culture dépend la possibilité d’un futur diagnostic prénatal enzymologique pour les grossesses suivantes. Il faut souligner qu’une activité normale de la chaîne respiratoire ne permet pas d’exclure totalement un déficit même si le tissu étudié exprime la maladie. Il est alors indispensable d’étudier un autre tissu. Quel que soit l’organe atteint, il est essentiel de pratiquer une biopsie de peau. L’apport de l’immunohistochimie Une étude immunohistochimique du muscle visualisant une répartition anormale des mitochondries (ragged red fibers par la coloration au trichrome de Gomori) ou révélant une activité faible de la succinate déshydrogénase ou de la cytochrome oxydase constitue également un critère diagnostique de maladie mitochondriale. Investigations moléculaires Si les études enzymologiques permettent de pointer quel est le complexe déficitaire de la chaîne respiratoire, elles ne permettent pas de pointer la sous-unité déficitaire. Il est donc souvent difficile de connaître l’origine génétique du déficit observé chez le patient. Une présentation clinique stéréotypée (cf. tableau 2), doit conduire à une étude moléculaire ciblée de l’ADNmt. Toutefois, on estime que seulement 5 à 10 % des maladies mitochondriales de l’enfant seraient liées à un défaut de l’ADNmt. Dans près de 90 % des cas, il s’agirait de mutations siégeant dans des gènes nucléaires codant pour des sous-unités de la chaîne respiratoire ou de mutations dans des gènes participant à la mise en place et au contrôle de la chaîne respiratoire et de l’ADNmt. Ainsi, l’hétérogénéité génétique des maladies mitochondriales est considérable, rendant souvent difficile l’identification de l’anomalie moléculaire à l’origine de la maladie. Il est souvent difficile de connaître l’origine génétique du déficit observé. Conclusion Si un déficit de la chaîne respiratoire doit être évoqué chez des patients présentant une atteinte inexpliquée, neuro-musculaire ou non, associant des organes apparemment sans relation, un retard psychomoteur peut être le signe révélateur d’une maladie mitochondriale. Il est donc indispensable de pousser les investigations cliniques, métaboliques, radiologiques qui permettront d’orienter vers le diagnostic de cytopathie mitochondriale.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :