Publié le 27 nov 2006Lecture 16 min
Les syndromes neurologiques paranéoplasiques : un diagnostic souvent difficile
Jean-Philippe CAMDESSANCHÉ, Jean-Christophe ANTOINE Hôpital Bellevue, CHU de Saint-Étienne
Les syndromes neurologiques paranéoplasiques sont rares mais doivent être reconnus car ils peuvent conduire à la découverte précoce d’un cancer et de ce fait à son traitement rapide. La mise en évidence des anticorps onconeuraux qui témoignent d’une physiopathologie auto-immune croisée entre le système nerveux et la tumeur a permis un grand progrès dans le diagnostic et la compréhension de ces syndromes.
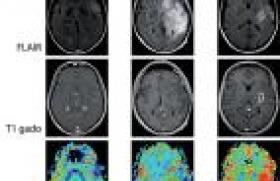
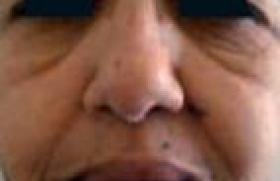
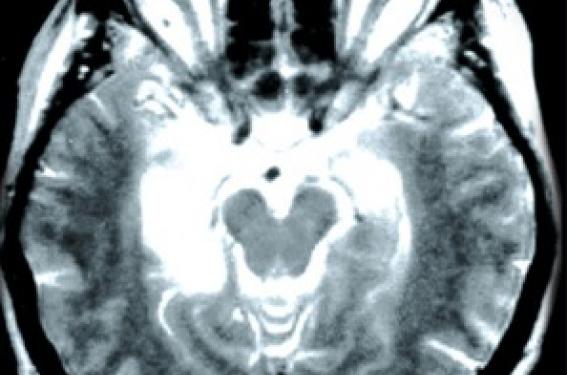
Un syndrome neurologique paranéoplasique (SNP) correspond à une atteinte du système nerveux dans un contexte de cancer systémique, en l’absence de complication iatrogène, infectieuse, métabolique, carentielle, métastatique ou d'infiltration de contiguïté du système nerveux par ce cancer (1). Les SNP sont rares ; leur fréquence a été estimée à 1 % des patients porteurs d’une tumeur maligne (2). Plusieurs mécanismes sont impliqués dans la physiopathologie de ces syndromes. Certains dépendent de troubles de la coagulation et de processus vasculaires. D’autres sont liés à la présence d’une gammapathie monoclonale. D’autres enfin mettent en jeu des mécanismes immuns et en particulier une réaction impliquant des antigènes dits onconeuraux car communs à la tumeur et au système nerveux. Ainsi, dans ce groupe, une réaction auto-immune primitivement antitumorale atteindrait secondairement le système nerveux (3). Les vingt dernières années ont vu le bouleversement des connaissances sur les SNP. La découverte des anticorps onconeuraux a permis d’améliorer les connaissances sur la physiopathologie de ces affections, les moyens du diagnostic et les stratégies thérapeutiques. Ceci a abouti en 2004, à la publication par un groupe de spécialistes européens, de critères diagnostiques pour les SNP (4). Confronté à un patient suspect de SNP, le clinicien est souvent conduit à se poser différentes questions. Le syndrome neurologique que j’observe est-il paranéoplasique et quels critères diagnostiques dois-je retenir ? Quelle est la valeur diagnostique des anticorps onconeuraux et sont-ils obligatoires pour le diagnostic ? Une fois le syndrome identifié, comment rechercher la tumeur et quel traitement proposer ? Par convention, ne seront pas abordées ici les neuropathies associées aux gammapathies monoclonales malignes, les complications vasculaires cérébrales du cancer et la myasthénie avec thymome. Poser le diagnostic de syndrome neurologique paranéoplasique La liste des affections neurologiques potentiellement paranéoplasiques est vaste (5). Les SNP impliquent toutes les structures neurologiques (système nerveux central, système nerveux périphérique, jonction neuromusculaire, muscle) et plusieurs syndromes même si l’un prédomine, peuvent s’intriquer chez un même patient (tableau 1). Les critères proposés par Graus et son équipe, résumés dans le tableau 2, prennent en compte le fait que les troubles neurologiques correspondent à un syndrome paranéoplasique classique ou non, la présence d’un anticorps onconeural bien caractérisé ou non, la survenue d’un cancer dans un délai raisonnable défini par rapport au début des signes cliniques et enfin une amélioration éventuelle du trouble neurologique avec le traitement de la tumeur. Selon ces critères, sont considérés comme étant des syndromes neurologiques paranéoplasiques classiques l’encéphalomyélite, l’encéphalite limbique, la dégénérescence cérébelleuse, l’opsomyoclonus, la neuronopathie sensitive subaiguë de Denny-Brown, la pseudo-occlusion digestive, le syndrome de Lambert-Eaton et la dermatopolymyosite (4). L’encéphalomyélite paranéoplasique C’est une atteinte multifocale du système nerveux central comportant une perte neuronale, des infiltrats inflammatoires perivasculaires et une gliose réactionnelle. Elle est rarement confinée au système nerveux central et s’associe très fréquemment à une neuronopathie sensitive et à des troubles dysautonomiques réalisant alors une encéphalo-myéloneuropathie. Le cancer le plus fréquemment rencontré est le cancer à petites cellules du poumon (CPCP). Le tableau clinique dépend du siège privilégié de l’atteinte et peut comporter des troubles de mémoire, une épilepsie, un syndrome cérébelleux, une rhombencéphalite, un syndrome d’atteinte du motoneurone exceptionnellement isolé. La dysautonomie peut comprendre une hypotension orthostatique, des troubles vaso- et sudomoteurs, des anomalies pupillaires et de plus rares troubles du rythme cardiaque. Une pseudo-occlusion digestive peut aussi être associée. Le liquide cérébrospinal est souvent inflammatoire avec une lymphocytose et une hyperproteinorachie modérée et parfois un profil électrophorétique oligoclonal (5). L’encéphalite limbique Elle correspond à une atteinte des structures temporo-mésiales. Son installation est volontierssubaiguë mais elle peut être plus rapide ou au contraire insidieuse. Elle associe anxiété, dépression, troubles du caractère mais comporte surtout un trouble de la mémoire antérograde avec oubli au fur et à mesure. D’autres manifestations sont possibles telles qu’une agitation, une confusion, des hallucinations et des crises d’épilepsie partielles ou généralisées. Le CPCP est le plus souvent associé avant le carcinome du testicule, la maladie de Hodgkin ou le thymome. Le liquide cérébrospinal est anormal comme dans l’encéphalomyélite. La présence d’anomalies radiologiques est nécessaire au diagnostic et correspond en IRM à un hypersignal T2 uni ou bilatéral des lobes temporaux internes (photo 1) et/ou au PET-scanner FDG à une hyperfixation du traceur au même endroit. L’EEG, quoique aspécifique, peut objectiver un ralentissement ou des décharges temporales. L’évolution qui se fait vers un état de démence peut parfois s’améliorer après traitement du cancer (5). Photo 1. Encéphalite limbique. IRM T2, coupe axiale, hypersignal temporal interne bilatéral asymétrique plus marqué à droite. La dégénérescence cérébelleuse Elle est responsable de l’apparition aiguë ou subaiguë d’un syndrome cérébelleux massif, bilatéral, statique et cinétique avec trouble de la marche et dysarthrie. Des vertiges et un nystagmus peuvent être présents. Les cancers classiquement associés sont le CPCP, les cancers gynécologiques (ovaire, utérus, sein) et la maladie de Hodgkin. Le liquide cérébrospinal est aussi volontiers inflammatoire pendant la phase initiale du tableau. L’imagerie précoce est normale, mais peut secondairement objectiver une atrophie (5). L’opsomyoclonus L’opsoclonus correspond à des mouvements oculaires involontaires, multidirectionnels déclanchés ou aggravés par la fixation ou les mouvements de poursuite. Il s’associe fréquemment à des myoclonies des membres. Il est plus souvent non paranéoplasique (virus, toxique, traumatique, ischémie cérébrale, etc.). Quand il est paranéoplasique, l’opsomyoclonus est rarement isolé mais se rencontre plutôt en association avec une atteinte cérébelleuse et/ou vestibulaire centrale. Son évolution est subaiguë et fluctuante. Il précède volontiers la découverte du cancer. Une amélioration spontanée est possible. Le LCR peut être inflammatoire. L’imagerie cérébrale ne montre pas d’anomalie particulière. Chez l’enfant, l’opsomyoclonus révèle un neuroblastome dans 2 à 7 % des cas. Chez l’adulte, il est volontiers associé à un cancer bronchique ou mammaire (5). La neuronopathie sensitive subaiguë Décrite par Denny-Brown en 1948, elle correspond à la destruction des neurones sensitifs dans les ganglions rachidiens postérieurs. Subaiguë, elle est classiquement asymétrique, distale et douloureuse. Elle débute volontiers aux membres supérieurs mais atteint au décours de la maladie les quatre membres et parfois le tronc et la face. Elle concerne toutes les modalités sensitives mais domine sur les grosses fibres. Elle associe douleurs, paresthésies, hypoesthésie et ataxie. L’importance de la déafférentation sensitive peut aboutir à des mouvements anormaux pseudo-choréo-athétosiques. Les réflexes ostéotendineux sont en général faibles ou abolis. Elle est fréquemment associée au CPCP. L’électrophysiologie objective une diminution ou une disparition des réponses sensitives. L’évolution irrémédiable aboutit à une grabatisation rapide du sujet qui fait souvent le pronostic de la maladie, plus que le cancer lui-même (5). La pseudo-occlusion digestive Secondaire à l’atteinte des plexus myentériques, elle s’exprime sous la forme d’une paralysie digestive plus ou moins étendue responsable d’un tableau occlusif avec nausées et vomissements. Dans certains cas, elle peut être isolée ou inaugurer l’histoire paranéoplasique (5). Le syndrome de Lambert-Eaton Il résulte d’un blocage présynaptique de la libération de l’acétylcholine des neurones moteurs et végétatifs périphériques par des anticorps dirigés contre les canaux calciques voltage-dépendants. Il correspond à un SNP dans environ 6 cas sur 10 et est associé à un CPCP dans 85 % des cas. Les patients présentent un déficit moteur progressif prédominant sur la ceinture pelvienne, une diminution ou une abolition des réflexes ostéotendineux et une atteinte végétative s’exprimant par une bouche sèche, une constipation ou une impuissance. L’électroneuromyogramme permet le diagnostic en montrant le blocage présynaptique au travers d’un phénomène de potentiation qui correspond à l’augmentation de l’amplitude du potentiel global d’action motrice supérieur à 100 % dans les suites d’un effort bref (photo 2) ou d’une stimulation à haute fréquence moins utilisée actuellement. À basse fréquence, un décrément sans cupule peut être observé. Le diagnostic peut être rendu difficile par l’intrication aux autres SNP. En cas de doute, sa recherche doit être systématique (5). Photo 2. Syndrome de Lambert-Eaton. Potentiation par un effort bref (10 secondes). Enregistrement réalisé par électrode de surface sur l’abductor pollicis brevis droit après stimulation du nerf médian au poignet avant et juste après l’effort. Gain d’amplitude de 122 %. La dermatopolymyosite Elle associe un érythro-œdème prédominant sur les zones découvertes (photo 3) et des papules de Gottron sur les articulations interphalangiennes. Le déficit musculaire est plus marqué au niveau des ceintures. Les muscles sont douloureux. Si la dermatopolymyosite n’est pas systématiquement paranéoplasique, l’occurrence d’un cancer est multipliée par six en sa présence pour atteindre 15 à 30 % des cas dans certaines études. Il s’agit en général d’un cancer du poumon, du col de l’utérus, de l’ovaire, d’un cancer digestif ou du pancréas5. L’étude histologique du muscle permet d’étayer le diagnostic clinique. Les anticorps onconeuraux La première description d’un auto-anticorps au cours d’un SNP remonte à 1965, mais c’est depuis les années 80 que les anticorps onconeuraux ont été clairement identifiés. Ce terme est réservé aux anticorps qui s’observent de façon quasi constante avec un cancer et pour lesquels il a été démontré que l’antigène reconnu est conjointement exprimé par la tumeur et le système nerveux. Une douzaine d’anticorps onconeuraux a été décrite. Les anticorps bien caractérisés (anti-Hu, Yo, CV2, Ri, Ma2 et amphiphysine) sont définis par l’existence d’un marquage spécifique en immunohistochimie sur coupe de tissu cérébral, la possibilité de les tester en Western blot avec la protéine recombinante correspondante et enfin leur validation par plusieurs équipes spécialisées (photo 4). Photo 4. Caractérisation des anticorps onconeuraux. Immunohistochimie sur cervelet de rat et Western blot avec la protéine recombinante correspondante. Les autres anticorps non complètement caractérisés doivent faire l’objet d’études complémentaires pour répondre à ces critères4. Les anticorps onconeuraux s’associent à des syndromes paranéoplasiques et des cancers précis (tableau 3). La spécificité de l’anticorps peut être grande comme pour l’anticorps anti-Yo qui est presque toujours associé à la dégénérescence cérébelleuse et à un cancer gynécologique (6) ou l’anticorps anti-Hu associé au syndromeencéphalomyélite/neuronopathie sensitive et au CPCP (7). D’autres anticorps sont moins spécifiques d’un tableau clinique et d’un cancer. Pour un anticorps spécifique, si le cancer détecté est différent de celui attendu, comme deux tumeurs peuvent survenir simultanément chez un même patient, il convient de rechercher activement la tumeur habituellement associée à l’anticorps. L’association de deux anticorps est possible. Cela est particulièrement vrai avec le CPCP où il n’est pas rare d’observer chez un même patient un anticorps anti-Hu et un anticorps anti-CV2 ou amphiphysine, parfois un anticorps anti-Ri. Ces associations représentent jusqu’à 30 % des cas8. À l’inverse, l’anticorps anti-Yo n’est qu’exceptionnellement associé à un autre anticorps. Le tableau 3 montre aussi qu’un même SNP peut survenir avec différents types d’anticorps. En pratique, le laboratoire qui fera la recherche d’anticorps onconeuraux doit pratiquer un screening d’ensemble. Les SNP classiques ne comportent pas toujours d’anticorps onconeuraux (9). Environ 40 % des dégénérescences cérébelleuses et des encéphalites limbiques et 20 % des neuropathies sensitives paranéoplasiques surviennent sans anticorps. Dans la majorité de ces cas, la tumeur sous-jacente est un CPCP. Existe-t-il des anticorps non encore identifiés ou la réponse immunitaire n’implique -t’elle pas l’immunité humorale ? Concrètement, l’absence de détection d’un anticorps onconeural n’exclut donc pas l’origine paranéoplasique d’un syndrome classique. Au total, il apparaît que pour un même tableau clinique et parfois pour une même tumeur (surtout le CPCP) plusieurs anticorps onconeuraux ou aucun anticorps peuvent être détectés (tableau 4). Environ 40 % des dégénérescences cérébelleuses et des encéphalites limbiques et 20 % des neuropathies sensitives paranéoplasiques surviennent sans anticorps. À l’inverse, malgré des recherches attentives et répétées et parfois même après autopsie, 2 à 5 % des patients ayant un anticorps onconeural ne présentent pas de cancer. Ces cas sont supposés liés à une destruction de la tumeur par la réponse immunitaire (3). Enfin 10 à 15 % des patients ayant un CPCP développent des anticorps anti-Hu ou CV2 sans SNP. Le titre de l’anticorps est en général inférieur à celui observé en cas de SNP. Cela doit être pris en compte avant d’admettre la nature paranéoplasique d’un syndrome neurologique inhabituel. Ce point a conduit certains auteurs à ne voir dans les anticorps onconeuraux que des marqueurs du cancer d’autant plus qu’il n’a jusqu’à présent pas été démontré qu’ils avaient un rôle directement pathogène (4). Le rôle de l’immunité dont témoignent les anticorps sur l’évolution du cancer est controversé. Certaines études chez des patients ayant un CPCP sans SNP tendent à montrer que le pronostic de la tumeur, ou sa réponse au traitement seraient meilleurs en cas d’association avec un titre faible d’anticorps anti-Hu. La recherche du cancer lors d’un syndrome neurologique paranéoplasique Dans 80 % des cas environ, le SNP précède l’apparition clinique du cancer (3). La recherche d’un cancer doit être systémtique en cas de SNP classique ou si un anticorps onconeural caractérisé est détecté. Hors de ce contexte, la discussion se fera au cas par cas. Le type de l’anticorps onconeural pourra orienter la conduite des examens complémentaires. La spécificité de l’anticorps anti-Yo pour les tumeurs gynécologiques est telle qu’en l’absence de mise en évidence d’un cancer du sein, il est proposé de pratiquer une laparotomie exploratrice à la recherche d’un cancer utéro-ovarien et une ovariectomie lorsque celui-ci n’est pas diagnostiqué par le bilan classique (6). La question d’une orchidectomie éventuellement bilatérale pourrait se poser de la même façon chez les hommes jeunes présentant un anticorps anti-Ma2 lorsque la tumeur n’est pas immédiatement apparente. Pour les autres anticorps ou les syndromes neurologiques sans anticorps, la tumeur le plus souvent en cause est le CPCP, mais d’autres tumeurs sont possibles. La recherche du cancer passe en premier lieu par un bilan d’imagerie classique (tomodensitométrie thoraco-abdomino-pelvienne ; échographie abdominale, pelvienne et testiculaire chez l’homme ; fibroscopie bronchique ; le cas échéant, examens endoscopiques digestifs). La spécificité de l’anticorps anti-Yo pour les tumeurs gynécologiques est telle qu’en l’absence de mise en évidence d’un cancer du sein, il est proposé de pratiquer une laparotomie exploratrice à la recherche d’un cancer utéro-ovarien.La rentabilité d’un tel bilan n’a été que peu étudiée. Dans certaines séries, chez des patients ayant un anticorps onconeural, une tumeur n’est mise en évidence que dans 65 à 75 % des cas lors d’un premier bilan. Le PET-scanner au 18 fluoro-desoxy-glucose (FDG) apparaît alors comme un examen clé car il a la capacité à mettre en évidence des tumeurs infra-centimétriques (Photo 5). Photo 5. PET-scanner au 18 FDG. Patiente aux antécédents de cancer de l’ovaire traité et considéré comme étant en rémission. Syndrome cérébelleux, anticorps anti-Yo. Examen permettant la découverte d’une image axillaire qui sera biopsiée et permettra le diagnostic de métastase du cancer ovarien. Sa sensibilité était de 83 % dans le travail de Younes-Mhenni et ses collaborateurs, chez des patients porteurs d’un anticorps onconeural (10). Sa spécificité est, en revanche, faible car le traceur peut fixer des tumeurs bénignes ou des lésions inflammatoires. En cas de bilan négatif, les recommandations actuelles proposent la répétition du bilan d’imagerie tous les six mois pendant deux ans. Au-delà, le risque de développer un cancer décroît significativement. En cas d’anticorps onconeural positif, cette recherche doit sans doute être maintenue dans le temps. Quels traitements pour les syndromes neurologiques paranéoplasiques ? À l’exception du syndrome de Lambert-Eaton, de la dermato-polymyosite, de la neuromyotonie et de la rétinopathie paranéoplasique qui sont susceptibles de répondre favorablement aux immunoglobulines intraveineuses ou aux corticoïdes (11), les autres SNP ont une évolution souvent dévastatrice et irréversible car rapidement accompagnés d’une dégénérescence neuronale. Sur la base de l’hypothèse d’une auto-immunité croisée entre la tumeur et le système nerveux, des thérapeutiques immunosuppressives et le traitement de la tumeur ont été proposés. Des publications de cas isolés traités en ouvert rapportent parfois une efficacité des traitements immunosuppresseurs tels que les corticoïdes, les échanges plasmatiques ou les immunoglobulines intraveineuses. L’encéphalite limbique et les neuropathies seraient les meilleurs candidates (3,5,11). Les grandes séries rétrospectives indiquent plutôt que le traitement le plus précoce et le plus complet du cancer serait un facteur indépendant du pronostic du SNP et en particulier de sa stabilisation (9). Les grandes séries rétrospectives indiquent plutôt que le traitement le plus précoce et le plus complet du cancer serait un facteur indépendant du pronostic du SNP et en particulier de sa stabilisation. À côté des traitements à visée étiopathogénique, des thérapeutiques symptomatiques non spécifiques peuvent être utiles (5). Cela inclut les antiépileptiques dans le traitement de l’encéphalite limbique, de la neuromyotonie (carbamazépine) et des douleurs neuropathiques. Les tricycliques sont aussi utiles pour le traitement de ces douleurs. Le syndrome de la personne raide peut être amélioré par le diazépam, le clonazépam ou le baclofen et l’opsoclonus par le clonazépam ou le propanolol. Enfin, le syndrome de Lambert-Eaton peut répondre à la 3,4-diaminopyridine et la pyridostigmine (5).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :