Publié le 19 nov 2007Lecture 18 min
Physiopathologie de la migraine : quelles avancées ?
Christian Lucas, Clinique neurologique, CHRU, Lille
Même s’il subsiste beaucoup de zones d’ombre dans la compréhension de la physiopathologie de la migraine, les techniques récentes d’imagerie, de biochimie et de génétique ont permis des avancées importantes qui ont donné naissance à de nouvelles approches thérapeutiques. Christian Lucas rassemble ici les différents éléments du puzzle dont nous disposons, sans oublier ceux qui manquent encore…
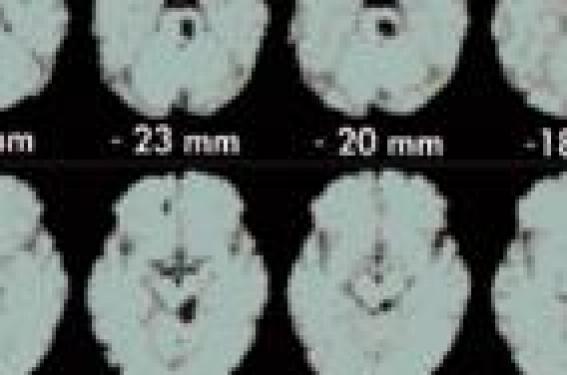
A l’instar de la plupart des affections neurologiques, la physiopathologie de la migraine a énormément évolué grâce notamment à l’imagerie fonctionnelle, la biochimie et la génétique. Il semble très loin le temps freudien où la migraine (sans doute du fait d’une prévalence plus importante chez la femme) était présumée purement psychique. Il y a eu ensuite, dans les années 1960, la théorie vasculaire avec l’alternance d’une vasoconstriction entraînant l’aura, suivie d’une vasodilatation des artères méningées entraînant la céphalée, théorie développée par Wolff (1), puis la théorie neuronale avec l’aura liée à la dépression corticale envahissante (spreading depression de Leao (2) mais qui n’expliquait pas les mécanismes de la céphalée. Ces théories furent ensuite battues en brèche par la théorie trigéminovasculaire (avec l’implication de possibles générateurs dans le tronc cérébral, des structures du trijumeau, des récepteurs sérotoninergiques 5HT1B/1D, qui allait donner naissance aux traitements de crise spécifiques que sont les triptans) avec la libération de différents neuropeptides proinflammatoires, dont le CGRP (calcitonin gene related peptide, avec actuellement des antagonistes des récepteurs du CGRP en phase III de développement dans le traitement de la crise) et le tout, soustendu par une susceptibilité génétique. Enfin, les modèles cliniques mais quasi-expérimentaux de la migraine hémiplégique familiale, affection autosomique dominante, font désormais considérer la migraine comme une possible maladie des canaux ioniques (« channelopathy ») permettant ainsi d’expliquer l’aspect paroxystique, évoluant par crises, de la maladie. Néanmoins, la ou les physiopathologies (un générateur et des effecteurs ou plusieurs modulateurs d’un seuil génétiquement déterminé, sans véritable générateur ?) de la migraine ou des migraines (une seule affection ou des entités distinctes ?) sont loin d’avoir livré tous leurs mystères… Le terrain migraineux L’hyperexcitabilité neuronale du migraineux Il existe de façon clairement démontrée une hyperexcitabilité neuronale dans la migraine, notamment du cortex occipital avec une pauvreté de la neurotransmission inhibitrice (faible densité en récepteurs GABA), une richesse de la neurotransmission excitatrice (densité très élevée en récepteurs NMDA) et une synchronisation fonctionnelle élevée (densité neuronale élevée et haut niveau d’architecture fonctionnelle, densité élevée en jonctions GAP, faible densité relative en astrocytes). Il a été ainsi démontré que les migraineux ont un seuil intercritique d’excitabilité du cortex occipital par stimulation magnétique transcrânienne plus bas que celui des témoins (3). Les migraineux ont également, lors d’enregistrements de potentiels évoqués multimodaux, un défaut d’habituation, c'est-à-dire de diminution de l’amplitude des potentiels normalement observée lorsqu’on répète les trains de stimulations. Les migraineux ont un seuil intercritique d’excitabilité du cortex occipital par stimulation magnétique transcrânienne plus bas. Gènes et migraine De tout temps, la migraine est apparue avoir un déterminisme familial avec de très fréquents antécédents familiaux migraineux retrouvés lorsque l’anamnèse est bien conduite. D’où l’idée d’une affection polygénique. C’est le modèle quasi-expérimental des migraines hémiplégiques familiales qui a permis d’éclairer d’un jour nouveau, non seulement la génétique mais aussi la physiopathologie de la migraine. En effet, l’hyperexcitabilité dans la migraine serait due à une libération excessive de glutamate avec un substratum génétique comme cela a pu être démontré clairement dans la migraine hémiplégique familiale avec mutation du gène CACNA1A (codant pour la sous-unité a1 formant le pore des canaux calciques voltages-dépendants de type P/Q) ou du gène ATP1A2 (codant pour la sous-unité a2 de la Na+/K+ATPase). Ces mutations entraînent sur les neurones ou les astrocytes, où ces canaux sont exprimés, une modification de fonction (gain ou perte) qui est susceptible d’entraîner à son tour une dépression corticale envahissante par l’augmentation de libération de glutamate. Il est vraisemblable, mais non prouvé à ce jour, que différents gènes sont impliqués dans la migraine plus « commune » que la migraine hémiplégique familiale, de sorte que la migraine est très probablement une canalopathie. Facteurs déclenchants des crises Les facteurs déclenchants les crises sont très divers et variés d’un patient à l’autre et, même parfois chez un même patient, avec au premier rang les facteurs psychologiques (stress, levée de stress, etc.), les facteurs hormonaux (règles), les facteurs alimentaires, etc. Le point commun entre tous ces facteurs d’allure très disparates est in fine d’être des facteurs modulant la neurochimie cérébrale via, par exemple, les voies noradrénergiques ou le cortex orbitofrontal. Ces facteurs sont vraisemblablement des facteurs de modulation chez des individus génétiquement prédisposés (« seuil migrainogène ») sur les « générateurs » de la migraine, qu’ils soient dans le tronc cérébral et/ou dans le cortex. Générateurs de la migraine : tronc cérébral ou cortex ? Différents noyaux du tronc cérébral (4) sont impliqués : chez 9 migraineux sans aura en phase céphalalgique, des augmentations de débit sanguin cérébral dans différentes régions du tronc cérébral, connues pour être activées dans la douleur, ont été identifiées avec également l’implication de la substance grise périaqueducale à proximité du locus coeruleus et du raphé, zones à projection sérotoninergique et noradrénergique (figure 1). Figure 1. Possible générateur de la migraine dans le tronc cérébral avec hyperhémie persistante des structures du tronc cérébral après soulagement de la céphalée migraineuse par du sumatriptan. Cet hyperdébit a la particularité de persister après la disparition de la migraine sous sumatriptan, contrairement aux autres zones initialement impliquées. Plus récemment, l’équipe française de G. Géraud5 a montré en 2004 une persistance en TEP et IRM, après soulagement total de la céphalée migraineuse lors d’une crise, d’une activation dans le tronc cérébral (mesencéphale et partie haute du bulbe) et de l’hypothalamus suggérant que ces structures puissent être des générateurs de la crise. La persistance de cette activation pourrait peutêtre expliquer les phénomènes de récurrences des crises nécessitant au plan thérapeutique des triptans à demi-vie longue et/ou l’association d’emblée triptan-AINS. Il pourrait s’agir de zones-clés situées à l’interface entre systèmes excitateurs et systèmes inhibiteurs de la nociception céphalique. Une hyperexcitabilité des neurones thalamocorticaux et corticocorticaux est présente en période de crises. Une hyperexcitabilité des neurones thalamo-corticaux et cortico- corticaux est présente en période de crises (expliquant la phono-, la photo-, l’osmophobie, l’allodynie tactile et thermique). Nous avons fait état plus haut des phénomènes d’hyperexcitabilité du cortex, notamment occipital. La primauté du tronc cérébral ou du cortex comme « générateur » de la crise migraineuse n’est pas établie à ce jour, sachant que les deux mécanismes sont loin d’être antithétiques et que ces structures pourraient avoir un rôle permissif sur le déclenchement de la crise sans être le(s) générateur(s) princeps. Mécanismes de l’aura migraineuse La dépression corticale envahissante ou « spreading depression » L’aura migraineuse constitue une manifestation neurologique transitoire survenant chez 10 à 15 % des migraineux, se développant en plus de 4 minutes, durant habituellement moins d’une heure et précédant le plus souvent la céphalée. Elle se manifeste par la survenue de manifestations visuelles puis éventuellement sensitives ou phasiques, et/ou motrices. L’aura migraineuse a longtemps été considérée comme le résultat d’une vasoconstriction primitive transitoire, la céphalée étant secondaire à une vasodilatation de rebond provoquant l’activation de nocicepteurs périvasculaires (1). La responsabilité de mécanismes neuronaux dans la migraine avait déjà été évoquée devant l’analogie entre l’aura et la dépression envahissante décrite par Leao en 1944 (2). La dépression corticale envahissante (DCE) correspond à une vague de dépolarisation neuronale et gliale, suivie d’une dépression neuronale de longue durée, observée expérimentalement après application de potassium sur les cortex de rats. Ce phénomène électrique est contemporain d’une vague d’hypoperfusion puis, quelques heures plus tard, d’une hyperperfusion cérébrale. La DCE progresse lentement à la surface du cortex puis est arrêtée par un sillon ou une scissure. La DCE induit une hyperactivité neuronale initiale avec la libération massive de K+ et de glutamate, suivie d’une entrée massive de Na+ et de Ca++ dans les neurones et les astrocytes. Les astrocytes, en situation d’hypofonctionnement, sont alors incapables de protéger le neurone du glutamate et du K+ qui sont en excès dans le milieu extracellulaire, avec pérennisation de la dépolarisation neuronale. Le glutamate joue un rôle essentiel avec blocage de la DCE par les antagonistes des récepteurs NMDA. Chez l’homme, le rôle de la DCE est resté longtemps controversé du fait de la rareté de déclenchement des DCE et des difficultés à interpréter les phénomènes électromagnétiques observés. Grâce à l’utilisation de stimulations visuelles répétées prolongées en IRM fonctionnelle (IRMf) par effet BOLD, Cao et coll. (6) ont pu démontrer que des zones cérébrales passaient d’un état de réponse à la stimulation visuelle vers un état de non-réponse selon un « front de dé-activation », se propageant avec le temps le long de la scissure calcarine à une vitesse de 3-6 mm/min. Ces données ont été confirmées plus récemment lors de l’étude d’auras migraineuses spontanées réalisée par l’équipe de Hadjikhani (7) (figure 2 a et b). Figure 2 (a) . Illustration graphique par Lashley de la « marche migraineuse » lors d’une aura visuelle avec hypothèse d’un phénomène d’une dépolarisation corticale envahissante de part l’organisation rétinotopique du cortex occipital. Figure 2 (b). La DCE a pu formellement être démontrée chez l’homme par Hadjikani en 2001 en IRM fonctionnelle avec effet BOLD lors de l’enregistrement d’auras spontanées. Ces phénomènes ne semblent toutefois pas spécifiques à l’aura, puisqu’ils peuvent parfois s’observer au cours de crises migraineuses sans aura (6). La dépression corticale envahissante semble donc bien être à l’origine de l’aura migraineuse, et est vraisemblablement impliquée dans la migraine sans aura avec soit une DCE survenant dans des zones silencieuses ou avec perturbation de l’excitabilité corticale mais sans traduction clinique. Ceci pose la question d’une seule et même maladie migraineuse, sans et avec aura, et la question d’une physiopathologie monoforme ou pas. La dépression corticale envahissante semble bien être à l’origine de l’aura migraineuse. Mécanismes de la céphalée migraineuse Système trigémino-vasculaire et inflammation neurogène L’innervation nociceptive des vaisseaux méningés hémisphériques est faite par des fibres issues de la branche ophtalmique du trijumeau et celle des vaisseaux de la fosse postérieure par des fibres issues de la racine C2. Les deux systèmes interconnectés forment le complexe trigéminocervical. C’est l’activation de ce complexe qui entraîne la céphalée, notamment par l’activation des terminaisons présynaptiques périvasculaires des neurones trigéminés avec la libération de neuropeptides vaso-actifs (CGRP, peptide Y, substance P), à l’origine d’un phénomène d’inflammation neurogène (extravasation des protéines plasmatiques, vasodilatation et auto-entretien de l’activation neuronale par une « soupe inflam matoire » périvasculaire) (figure 3). Figure 3. Stimulation du système trigéminovasculaire lors d’une crise de migraine avec influx ortho- et antidromique entraînant une vasodilatation et une libération de substances pro-inflammatoires (CGRP, substance P, bradykinine), avec ensuite une stimulation du tronc cérébral et du système nerveux autonome puis une conduction des informations nociceptives jusqu’au thalamus et au cortex, accompagnée d’une sensation consciente de douleur. À noter, la présence de récepteurs 5-HT neuronaux et méningés, sites d’action des triptans. Selon la théorie neurotrigéminovasculaire de Moschowitz (8), la stimulation de nocicepteurs périvasculaires de la dure-mère et des méninges et/ou les nocicepteurs cervicaux active les neurones du noyau trigéminal caudal. Les influx nerveux générés seraient relayés de façon orthodromique jusqu’au thalamus puis au cortex, et permettraient l’élaboration de la perception consciente de douleur, tandis que l’activation antidromique des fibres trigéminales amyéliniques entraîne une libération de neuropeptides (SP, CGRP, neurokinine A). Les relations entre la DCE, l’aura et la céphalée migraineuse restent incertaines. Des travaux expérimentaux8 sont en faveur du déclenchement des céphalées par la DCE survenant en phase d’aura. Ils montrent que la répétition de la DCE induit une augmentation de l’activité neuronale dans le noyau caudal du trijumeau, élément-clé dans la genèse de la sensation douloureuse. Sensibilisation périphérique et centrale : le problème de l’allodynie Certains éléments cliniques font penser à des phénomènes de sensibilisation périphérique et centrale dans la migraine, avec notamment a toux ou la position penchée en avant qui peut aggraver la céphalée lors d’une crise ou la réveiller quelques heures après son terme, l’hyperesthésie du scalp avec la diminution du seuil douloureux, plus accentué du côté douloureux au cours d’une crise, et avec parfois la persistance jusqu’à quelques jours après son terme. À partir d’observations d’allodynie mécanique de degré variable chez différents migraineux, Burstein et coll. (9) ont émis l’hypothèse d’une sensibilisation de neurones trigéminothalamiques liée à la répétition des crises migraineuses. L’existence d’une allodynie témoignant d’une sensibilisation centrale apparaissant pendant ou au décours d’une crise migraineuse est de plus en plus discutée comme un élémentclé de la physiopathologie de la crise migraineuse, au point d’être avancée comme justifiant l’utilisation précoce des triptans au cours de la crise migraineuse. Mathew et coll. (10) ont considéré ce concept dans une approche résolument clinique. Ils ont interrogé rétrospectivement près de 300 pa tients à l’aide d’un questionnaire semi-structuré avec évaluation de la fréquence clinique de l’allodynie, et ils ont recherché des éléments cliniques corrélés à sa présence. Plus de la moitié (53,3 %) des patients ont fait état de ce phénomène au cours de leurs crises migraineuses. Cette allodynie était décrite comme essentiellement céphalique (84,7 %), ipsilatérale à l’hémicrâne le plus douloureux, et survenant le plus souvent à l’acmé de la crise. Une corrélation entre la présence d’une allodynie et l’ancienneté de la maladie migraineuse, ainsi qu’entre la présence d’une allodynie et la fréquence des crises a pu être mise en évidence. Ces derniers éléments sont importants à considérer car ils resituent les implications de cette allodynie bien au-delà de la justification d’une utilisation précoce des triptans. En effet, ils plaident en faveur d’une sensibilisation centrale comme substrat physiopathologique de la migraine chronique ce qui, dans le futur, pourrait conduire à une re cherche plus systématique de ce phénomène et à une modification de nos attitudes thérapeutiques en termes de traitement préventif. Ils restent cependant à confirmer ces résultats de façon prospective, ce qui permettrait de mieux évaluer les éléments cliniques corrélés à la présence de l’allodynie et d’établir la variabilité intra-individuelle de cette manifestation clinique. Certains éléments cliniques font penser à des phénomènes de sensibilisation périphérique et centrale dans la migraine. Données nouvelles sur la physiopathologie de la migraine Au cours de la session consacrée aux neurosciences cliniques lors du congrès de l’American Academy of Neurology 2007 à Boston, Mosckowitz11 a présenté une synthèse des données de la littérature sur la migraine, les gènes, les hormones et les canaux ioniques. Le pivot central de la communication reposait sur la DCE, ses rapports avec les dysfonctionnements des canaux calciques et des pompes sous-tendus par des mutations génétiques, et sur les applications en ce qui concerne les traitements de fond. Ces traitements de fond sont efficaces à la fois sur les migraines avec et sans aura, corroborant l'hypothèse d'une expression silencieuse de la DCE au cours des migraines sans aura. Cliniquement, la DCE, retrouvée chez tous les mammifères, se propage à une vitesse de 3 à 5 mm/min. Elle peut être déclenchée soit par le dépôt de KCL soit par l’ouabaïne, le glutamate et l'hypoxie. Elle est corrélée à une période brève d'hyperperfusion suivie par une hypoperfusion. Une accumulation locale de K+ dans l’espace extracellulaire pourrait être à l'origine d'une dépolarisation de la membrane présynaptique, d'une libération d’ions calcium et d'un gonflement des neurones post synaptiques. La traduction clinique de ce dysfonctionnement ionique est illustrée par la migraine hémiplégique familiale (MHF), pour laquelle 3 types sont, à l'heure actuelle, connus. Cette maladie représente un lien entre l'implication des gènes et la DCE. Au cours de cette pathologie, les crises sont déclenchées par le stress, l'effort, un traumatisme même minime. Les crises se manifestent par des migraines avec aura et le nombre de crises serait plus important chez la femme. La MHF est en rapport avec une mutation sur le gène CACNA1A pour le type 1 et avec une mutation sur la sousunité a2 de la pompe ATPase dépendante pour le type 2. Les variations de susceptibilité de la DCE entre les deux sexes ont été testées chez un modèle de souris (R122QK1), avec l'hypothèse que l'imprégnation hormonale pouvait expliquer une proportion plus importante de crises chez la femme. Il a été démontré que l'ovariectomie entraînait chez la souris une diminution de la survenue de la DCE. Les mêmes résultats ont été reproduits chez la souris ménopausée. Il existe donc chez l'animal des preuves d'une relation entre les hormones ovariennes et la DCE. Les traitements de fond pourraient agir en supprimant la DCE. Dans le travail rapporté par Ayata en 2006 (12, 5) substances ont été essayées (valproate, topiramate, bêtabloquants, amitriptylline et méthylgergide) chez l'animal, prouvant avec un délai de plusieurs semaines l'efficacité de ces substances tout en diminuant la DCE. Malgré cette avance physiopathogénique, il n'y a pas à ce jour d'explication pour commenter l'efficacité de drogues aussi différentes en tant que traitement de fond. Enfin, les travaux très récents de Takano sur la NADH (nicotinamide adénine dinucléotide), substance responsable du transfert d'énergie dans la chaîne mitochondriale et mesurée par techniques fluorométriques, ont démontré que l'hypoxie était un facteur-clé du déclenchement de la DCE. La dépression corticale propagée induite chez le rat par l’application de potassium à la surface corticale provoque d’importantes modifications structurelles et fonctionnelles de la barrière hémato-encéphalique en activant la métallo protéinase MMP9. La concentration intracérébrale homolatérale en MMP9 augmente dès la troisième heure après induction de la dépression corticale propagée, culmine à 24 heures et persiste jusqu’à 48 heures. L’activité lytique spécifique des métalloprotéinases est détectable précocement au niveau des vaisseaux corticaux (< 3 heures), altère ensuite l’arachnoïde puis s’exerce significativement en moins de 24 heures au niveau du cortex piriforme, activité non détectable en cas d’application conjointe d’un inhibiteur de la MMP9. Les protéines endothéliales et ceux de la membrane basale sont altérées environ 3 heures après le début de la DCE, altération qui s’accompagne d’un oedème cérébral et d’une extravasation plasmatique. Aucun de ces phénomènes ne survient chez la souris knock-out dépourvue d’expression de la MMP9. La dépression corticale propagée induit ainsi une cascade biochimique conduisant à la rupture de la barrière hémato-encéphalique par un mécanisme activateur dépendant de la métalloprotéinase MMP9. Ces données suggèrent de possibles risques lésionnels liés à la survenue d’une aura migraineuse (hypersignaux de substance blanche ?). Elles ouvrent également la voie à de nouvelles pistes thérapeutiques : certains inhibiteurs de l’activité métalloprotéinase pourraient être utilisés. Cette « lecture » remarquable de Mosckowitz permet de mieux articuler les différents éléments du puzzle de la physiopathologie de la migraine (figure 4) avec un scoop sur l’implication de la NADH, qui constituerait peut-être une piste pour la discussion physiopathologique des hypersignaux de la substance blanche dans la migraine. Figure 4. Physiopathologie présumée de la migraine (CERP : Calatoningene selected peptide, MPP9 : métalloprotéinase de type 9).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :




