Publié le 30 jan 2013Lecture 17 min
Les antiplaquettaires dans la prévention secondaire de l’accident vasculaire cérébral ischémique
S. RICHARD*, M. TOUSSAINT-HACQUARD**, J.-C. LACOUR*, P.-A. BAILLOT***, X. DUCROCQ* , *Service de neurologie, hôpital central, CHU de Nancy **Service d’hématologie biologique-hémostase médicale, hôpitaux de Brabois, CHU de Nancy, Vandoeuvre-les-Nancy ***Se
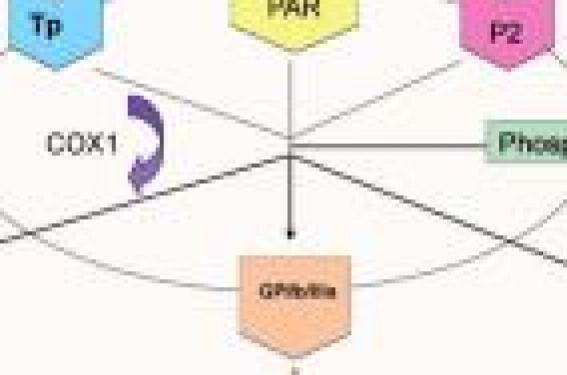
L’emploi des antiplaquettaires est une des mesures les plus importantes dans la prévention secondaire de l’accident vasculaire cérébral (AVC) ischémique. En inhibant l’activité plaquettaire, ils préviennent la formation du thrombus responsable de l’occlusion et de l’embolie artérielle.
L’aspirine, le clopidogrel et l’association aspirine-dipyridamole sont les seuls actuellement utilisés pour le cas particulier de la prévention secondaire de l’AVC ischémique, chacun représentant une voie différente d’inhibition plaquettaire. Des études ont comparé leur efficacité, ou même l’effet de leur association. La recherche fondamentale a montré, quant à elle, que l’inhibition plaquettaire pouvait être sujette à une variabilité interindividuelle conditionnée par des facteurs intrinsèques (polymorphisme génétique) ou extrinsèques (interactions médica menteuses). Une revue détaillant les différentes études cliniques qui guident aujourd’hui la prescription des antiplaquettaires, leur mode d’action et les facteurs de moindre réponse plaquettaire, pourrait aider le clinicien à mieux les utiliser. Les différentes voies d’inhibition plaquettaire La rupture de la plaque athéromateuse met à nu la matrice sousendothéliale contenant du collagène capable d’activer les plaquettes et donc de former un thrombus qui pourra soit réduire, soit occlure la lumière artérielle ou donner naissance à un embole pouvant être à l’origine d’une ischémie distale(1). L’activation plaquettaire comprend l’agrégation, mais aussi la synthèse de composés qui, en agissant sur différentes voies, l’entretiennent et la propagent (figure 1). Chacune de ces voies représente une cible pharmacologique d’inhibition (figure 2). La cible de l’aspirine est la cyclo-oxygénase (COX) 1, enzyme responsable de la production du plus puissant activateur plaquettaire : le thromboxane (TX) A2. L’activation des récepteurs P2 par l’ADP joue un rôle important dans l’activation plaquettaire. Les thiénopyridines, dont fait partie le clopidogrel, bloquent le récepteur P2Y12 de façon irréversible. Le clopidogrel est converti en métabolite actif, notamment par le cytochrome P4503A4, avant sa liaison avec P2Y12. Le dipyridamole inhibe la phosphodiestérase plaquettaire et donc la captation de l’adénosine, maintenant un taux élevé d’AMPc, avec diminution des concentrations de calcium intracellulaire et inhibition plaquettaire. Figure 1. Les différentes voies d’activation plaquettaire. Figure 2. Les différentes cibles d’inhibition plaquettaire. Chaque voie d’activation plaquettaire représente une cible pharmacologique, les antiplaquettaires ayant pour rôle l’inhibition de ces voies. Cependant, l’inhibition d’une seule voie peut-être insuffisante et l’activité plaquettaire maintenue. Les études cliniques en prévention secondaire de l’AVC ischémique Pour l’aspirine, il faut différencier deux phases dans la prévention secondaire de l’AVC ischémique. • La phase aiguë correspondant aux 7 premiers jours suivant l’AVC ischémique. L’aspirine est actuellement le seul antiplaquettaire utilisé à cette phase. Deux grandes études s’y sont intéressées : IST(2) et CAST(3) durant les 48 heures qui suivent l’événement, pour des doses de 160 et 300 mg d’aspirine. Ces études ont regroupé 60 000 patients et montré que l’administration d’aspirine pendant cette phase permet d’éviter 9 AVC ischémiques et décès pour 1 000 patients traités durant le premier mois. • Pour la prévention dite « au long terme », une métaanalyse(4) regroupant plus de 287 essais cliniques a montré que les antiplaquettaires (la majorité des patients étaient traités par aspirine) réduisaient approximativement de 23 % le risque relatif d’AVC ischémique, d’infarctus du myocarde et de mort vasculaire pour des patients atteints de maladies cardio- et cérébrovasculaires. Pour les thiénopyridines, le clopidogrel a succédé à la ticlopidine dont la tolérance hématologique a limité la prescription. L’étude CAPRIE(5) a comparé l’effet de clopidogrel (75 mg/j) à celui de l’aspirine (325 mg/j) en prévention secondaire d’un événement artériel ischémique chez près de 20 000 patients. Ceux traités par clopidogrel ont présenté un taux d’événements ischémiques (AVC ischémiques, infarctus du myocarde et mort vasculaires) de 5,32 % par an, contre 5,83 % par an pour les patients traités par aspirine. Le risque hémorragique (9,2 %) est le même que celui de l’aspirine, mais avec une meilleure tolérance digestive. Même si cette différence est significative, le bénéfice décrit dans cette étude avec clopidogrel reste modeste en comparaison de celui obtenu avec l’aspirine. Pour le dipyridamole, l’étude ESPS 2(6) a comparé l’effet de l’aspirine seule (50 mg/j) au dipyridamole seul (400 mg/j) et à l’association de ces 2 molécules versus placebo, en prévention secondaire de l’AVC ischémique, avec une durée moyenne de suivi proche de 2 ans. Le risque de récidive ischémique cérébrale et de mort vasculaire a été réduit de 13 % dans le groupe de patients traités par aspirine, de 15 % pour ceux traités par dipyridamole, mais surtout de 24 % pour ceux traités par l’association en comparaison au placebo. Ces résultats ont permis de montrer l’efficacité, en prévention secondaire de l’AVC ischémique, du dipyridamole, mais surtout d’une meilleure protection lorsqu’il est associé à l’aspirine. Ces conclusions ont été confirmées par l’étude ESPRIT(7) qui a comparé, en prévention secondaire de l’accident ischémique transitoire et de l’AVC ischémique mineur, l’effet de l’association de l’aspirine (75 mg/j) au dipyridamole (200 mg x 2/j) à celui de l’aspirine seule. Pour une durée moyenne de suivi de 3,5 ans, une diminution du risque absolu de 1 % par an du critère combiné mort vasculaire, AVC ischémique, infarctus du myocarde et complications hémorragiques majeures a été constatée pour l’association (13 % d’événements) comparativement au traitement par aspirine seul (16 % d’événements). Cependant, les abandons de traitement ont été plus fréquents dans le groupe traité par l’association en raison de la survenue de céphalées. L’effet du clopidogrel (75 mg/j) et de l’association aspirine (25 mg x 2/j) et du dipyridamole (200 mg x 2/j) a été comparé par l’étude PROFESS(8), qui a inclus plus de 20 000 patients. Il n’y a pas eu de différence significative pour les récidives d’AVC ischémiques, avec une fréquence de 9 % dans les deux groupes, ni pour le pronostic fonctionnel et l’impact cognitif après ces récurrences. L’effet de l’association du clopidogrel (75 mg/j) à l’aspirine (75 mg/j) a été comparé à du clopidogrel seul dans l’étude MATCH(9), qui a inclus plus de 7 500 patients après un événement cérébral ischémique. Après un suivi moyen de 18 mois, 15,7 % des patients traités par l’association ont présenté une récidive d’AVC ischémique, un infarctus du myocarde ou un décès d’origine vasculaire contre 16,7 % des patients traités par clopidogrel seul. Cette différence en faveur d’une meilleure protection par l’association, n’était pas statistiquement significative. De plus, ce léger bénéfice était annulé par une augmentation du nombre de saignements engageant le pronostic vital dans le groupe traité par l’association (2,6 %) en comparaison au groupe traité par clopidogrel seul (1,3 %). L’association est, en revanche, utilisée de façon courante dans le domaine cardiovasculaire en prévention des thromboses des stents coronariens et donc, par prolongation, dans le domaine neurovasculaire pour les traitements interventionnels des troncs supra-aortiques et des artères intracrâniennes où elle encadre le geste pendant plusieurs mois. Elle pourrait également être étudiée dans des cas très précis, comme pour les sténoses athéromateuses(10) ou, sur une durée limitée, à la phase aiguë de l’AVC ischémique. L’effet d’un antagoniste spécifique des récepteurs du TXA2, appelé S18886 ou terutroban, a été comparé à celui de l’aspirine seule, en prévention secondaire de l’AVC ischémique et de l’AIT, chez près de 20 000 patients dans l’étude PERFORM(11). Malheureusement, le terutroban n’a pas montré de supériorité par rapport à l’aspirine, avec 11 % de récidives d’AVC ischémiques, d’infarctus du myocarde et de mort vasculaire pour les deux groupes. Cependant, l’analyse des sous-groupes a montré que la survenue d’événements ischémiques artériels était significativement moins fréquente avec le terutroban pour des patients ayant présenté l’événement qualifiant sous aspirine. Cette molécule semblait donc utile pour des patients en « échec clinique de l’aspirine ». Tests d’exploration des fonctions plaquettaires Les nombreuses études citées ci-dessus ont montré que les récidives ischémiques sous antiplaquettaires après un AVC ischémique ne sont pas rares. Ceci a fait naître la notion de « résistance clinique aux antiplaquettaires ». Afin de comprendre les mécanismes de moindre réponse, l’étude de l’effet biologique des antiplaquettaires a été développée. Différents tests existent, mais sont encore peu utilisés dans la pratique courante, surtout neurovasculaires. La corrélation avec la clinique et entre les résultats des différents tests est encore mal établie. L’utilisation de ces méthodes de mesure de l’activité plaquettaire et de l’effet de l’inhibition des antiplaquettaires a permis de mettre en évidence une variabilité de réponse interindividuelle non dichotomique entre les patients, c’est pourquoi le terme « variabilité de réponse plaquettaire » doit être préféré à celui de « résistance » aux antiplaquettaires. L’agrégation photométrique par variation de transmission lumineuse est un test réalisé dans les laboratoires spécialisés, qui permet d’explorer le fonctionnement plaquettaire avec un plasma riche en plaquettes, anticoagulé avec du citrate, puis stimulé par différents agonistes (collagène, ADP, acide arachidonique). Figure 3. Agrégométrie photométrique, avec profil d’agrégation caractéristique d’un traitement par aspirine. Pour agrandir cliquez ici ou sur la figure L’agrégation est me surée par variation de transmission lumineuse et exprimée en pourcentage par rapport à un plasma pauvre en plaquettes. Les principaux antiplaquettaires induisent une thrombopathie caractéristique. L’aspirine inhibe fortement l’agrégation induite par l’acide arachidonique (figure 3) et l’agrégation dite secondaire (dépendant de la synthèse du TX par les plaquettes activées par l’ADP ou le collagène à faible concentration). La réponse aux fortes concentrations de collagène est préservée. Le clopidogrel inhibe fortement l’agrégation induite par l’ADP (figure 4). Cette méthode est maintenant considérée comme le « gold standard » dans la littérature. Plus récemment, un test fondé sur le principe de l’agrégation photométrique, mais réalisé en sang total et avec une cartouche de réactif spécialement dédiée à la réponse à l’aspirine, a été commercialisé (VerifyNow®, Accumetrics). Figure 4. Agrégométrie photométrique avec profil d’agrégation caractéristique d’un traitement par clopidogrel. Pour agrandir cliquez ici ou sur la figure. Ce test développé pour être réalisé au lit du malade, consiste à quantifier la capacité des plaquettes, après stimulation à l’acide arachidonique, à se lier à des billes couvertes de fibrinogène. La méthode de détection est basée sur la transmission d’un faisceau lumineux et le résultat est exprimé en ARU (Aspirin Reaction Units) avec un seuil fixé par le fabricant à 550 ARU, seuil au-delà duquel le patient peut être considéré comme mauvais répondeur. Ce test n’explore que la réponse à l’aspirine, sans être spécifique, sur la base de la réactivité à l’acide arachidonique. Parmi les autres tests fonctionnels qui peuvent être utilisés pour apprécier le retentissement biologique de l’aspirine, il existe des tests globaux d’exploration de l’hémostase primaire avec une importante variabilité interindividuelle de sensibilité : – temps de saignement in vivo ; – PFA-100 ex vivo qui consiste à mesurer avec du sang total, anticoagulé par du citrate, soumis à des contraintes de cisaillement, le temps d’occlusion d’un orifice au centre d’une membrane couverte d’un mélange de collagène/ADP ou de collagène/épinéphrine ; – mais aussi des tests permettant d’apprécier la réactivité plaquettaire par cytométrie en flux : mesure de l’expression membranaire de la P-sélectine notamment, qui est une glycoprotéine peu exprimée lorsque la plaquette est à l’état quiescent et surexprimée après activation plaquettaire et sécrétion de granules alpha.Des tests se proposent de mesurer la réponse à l’aspirine sans explorer la fonctionnalité plaquettaire, mais en testant sa capacité résiduelle de synthèse du TXA2 par dosage de ses métabolites, le TXA2 étant un composé très instable et rapidement métabolisé ; – mesure du TXB2 dans le sérum après stimulation maximale in vitro des plaquettes par la thrombine lors de la coagulation du sang total ; – mesure du 11-déhydro TXB2 urinaire reflétant l’ensemble du TX généré in vivo, quelles qu’en soient les sources (plaquettaires et extraplaquettaires) ; – analyse de la phosphorylation de VASP. La protéine VASP (Vasodilatator Stimulated Phosphoprotein) est une protéine ubiquitaire qui se trouve dans les plaquettes sous deux formes, l’une phosphorylée et l’autre non. Son pourcentage de phosphorylation est inversement proportionnel à l’activité du récepteur P2Y12, cible des thiénopyridines. Les variabilités de réponse aux antiplaquettaires C.M. Helgason et coll.(12) ont fait naître le concept dans le domaine neurovasculaire de « résistance à l’aspirine » en montrant des variabilités de réponse plaquettaire mesurée par agrégométrie photométrique, en fonction de la dose et de la durée de prescription. Puis d’autres travaux ont tenté de démontrer qu’une mauvaise réponse plaquettaire à l’aspirine, évaluée par des examens biologiques ex vivo, est associée à un risque important de survenue d’un épisode thrombotique artériel. Cependant, les tests utilisés sont différents selon les études. F.M. Gengo et coll.(13) ont mesuré par agrégométrie photométrique la réponse plaquettaire à l’aspirine d’une population de patients traités, depuis au moins 2 semaines, suite à un AVC ischémique. Parmi ceux qui avaient présenté une récidive, 66 % avaient une mauvaise réponse biologique. Le concept de résistance à l’aspirine reste donc très imprécis. Il inclut à la fois des épisodes thrombotiques artériels chez des patients traités par aspirine et des variabilités de réponse plaquettaire à l’aspirine mesurées par différents examens biologiques ex vivo différents. Pour les études citées, la définition de « résistance biologique » est très variable rendant ainsi les métaanalyses difficiles. La seule réalisée à ce jour(14), a montré une association positive entre les événements cliniques et les résultats des tests biologiques. De nombreux mécanismes physiopathologiques ont été incriminés pour la moindre réponse plaquettaire à l’aspirine. Mécanismes pharmacocinétiques Il s’agit du plus fréquent de ces mécanismes(15). Le défaut d’observance du traitement est la première cause à évoquer. La diminution de l’absorption digestive peut être incriminée pour les formes « gastro-résistantes » d’aspirine, non prescrites en France actuellement. Ces formes sont prescrites pour diminuer la toxicité gastrique de l’aspirine, cependant l’absorption a lieu dans l’intestin où le pH est plus élevé que dans l’estomac. L’aspirine est alors inactivée par désacétylation et moins soluble, donc moins absorbée. Le poids par augmentation du volume de distribution peut être un facteur de moindre réponse(16). La prise d’aspirine doit être quotidienne afin de prendre en compte le renouvellement plaquettaire(17). La prise concomitante d’anti-inflammatoire non stéroïdien (AINS) peut gêner l’action de l’aspirine sur la COX(18). L’AINS provoque une inhibition compétitive de la COX1 sur le même site que l’aspirine, mais le blocage, contrairement à l’aspirine, est réversible. L’aspirine, ayant une demi-vie très courte, n’est plus présente dans la circulation quand le site actif de la COX1 est libéré par l’AINS. L’une des solutions serait de prendre l’aspirine au moins 2 heures avant l’AINS. Mécanismes pharmacodynamiques dépendant de la production de TXA2 La diminution de sensibilité de la COX1 à l’aspirine représenterait un vrai mécanisme de moindre réponse. Le polymorphisme du gène de la COX1 a été largement étudié, dans l’hypothèse d’une moindre réponse due à une modification du site actif de l’enzyme. Mais les données de la littérature sont contradictoires(19). Le diabète, par glycation des protéines du site actif de la COX1, diminuerait l’affinité de l’aspirine(20). D’autres mécanismes agissent en produisant du TXA2 par d’autres sources que la COX1. Ainsi, la COX2 est une enzyme inductible, trouvée dans les cellules nucléées, dont les macrophages, les monocytes et les cellules endothéliales. Elle est induite par des stimuli inflammatoires. Elle est capable de produire des précurseurs du TXA2 malgré l’inhibition de la COX1 et synthétise d’autres composés comme les leucotriènes ou les isoprostanes favorisant l’agrégation plaquettaire(21). Son action délétère dans l’ischémie cérébrale a été montrée(22). Voies d’activation plaquettaire indépendantes de la COX1 et de la production de TXA2 L’hypertension artérielle(23), le tabagisme(24), l’hypercholestérolémie totale et le LDL cholestérol(25) diminuent la réponse plaquettaire à l’aspirine sans que les mécanismes physiopathologiques soient élucidés. Les isoprostanes naissent de l’oxydation des lipides membranaires par une voie indépendante des COX(26). Ces composés induisent une vasoconstriction et une augmentation de l’agrégabilité plaquettaire via les récepteurs du TXA2. Leur production peut être favorisée par le stress, le diabète, le tabac, l’hypercholestérolémie, l’hyperhomocystéinémie et le vieillissement. Des leucotriènes pourraient avoir la même action. L’interaction des plaquettes avec les érythrocytes et les leucocytes peut favoriser leur activation(27). Variabilité de réponse au clopidogrel Celle-ci serait beaucoup plus importante que celle constatée avec l’aspirine et pourtant beaucoup moins décrite dans le domaine neurologique. Les études sont surtout situées dans le domaine cardiovasculaire, où cette molécule est utilisée en association à l’aspirine dans la prévention des thromboses des stents coronariens. P. Fontana et coll.(28) ont administré à 96 volontaires sains, de façon quotidienne, 100 mg d’aspirine pendant 7 jours, puis après un « wash out » de 2 se maines, une première dose de 300 mg de clopidogrel, puis 75 mg par jour pendant 6 jours. Après le traitement par aspirine, 95 patients avaient des concentrations effondrées de TXB2 et une inhibition de l’agrégation induite par l’acide arachidonique. Un seul patient présentait une moindre réponse, qui était vaincue par augmentation des doses d’aspirine. Après traitement par clopidogrel, les résultats d’agrégométrie induite par l’ADP étaient disparates allant de 18 à 78 %. Le clopidogrel doit être converti en métabolite actif, notamment par les cytochromes P450 3A4 et 2C19 et l’estérase PON1(29). De nombreux médicaments, comme certains inhibiteurs de la pompe à protons(30), empruntent la même voie et peuvent interférer avec son métabolisme. Les polymorphismes de ces enzymes et des récepteurs P2Y12 sont également incriminés(31). Le dipyridamole étant utilisé, en prévention secondaire de l’AVC ischémique, exclusivement associé à l’aspirine, ses possibles variabilités d’inhibition plaquettaire n’ont jamais été décrites.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :