Publié le 27 jan 2009Lecture 9 min
Les malformations cérébelleuses. Une cause non négligeable de retard mental et d’encéphalopathie
L. BURGLEN, Hôpital Trousseau, Paris
Les malformations cérébelleuses sont un groupe d’affections d’origine fréquemment génétique. Le diagnostic positif repose sur l’IRM cérébrale, demandée devant une symptomatologie variable : grande hypotonie des premiers mois, troubles oculomoteurs, encéphalopathie, retard mental, ataxie. Il est important de tenter d’aboutir à un diagnostic étiologique précis qui sera essentiel pour le conseil génétique.
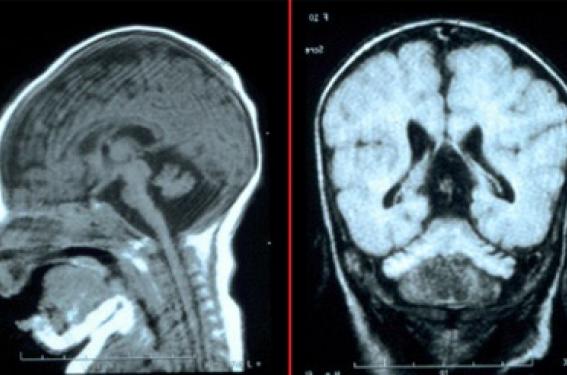
Les malformations du cervelet sont définies par l’existence d’une anomalie quantitative et/ou qualitative du cervelet (vermis et/ou hémisphères) présente à la naissance. Le diagnostic est fait soit en anténatal, soit en post-natal sur l’IRM cérébrale demandée devant une atteinte neurologique variable, mais en règle présente avant l’âge de 1 an. Ces malformations sont relativement fréquentes, toutes formes confondues, constituant une part non négligeable des encéphalopathies et retard mentaux de l’enfant. Elles constituent un groupe très hétérogène sur les plans clinique, radiologique et génétique. Le pronostic est difficile à établir lorsque l’anomalie est décelée en anténatal ou en période néonatale. Une malformation peu importante apparemment peut être responsable d’une encéphalopathie sévère. À l’inverse, dans de rares cas, une imagerie cérébelleuse très anormale peut coexister avec des formes cliniques peu sévères. Il est donc essentiel de tenter d’aboutir à un diagnostic étiologique précis. Une malformation peu importante apparemment peut être responsable d’une encéphalopathie sévère. Une origine génétique fréquente Pour les causes génétiques identifiées, peu de gènes sont connus. Il existe un risque pour les grossesses ultérieures, souvent sans possibilité de diagnostic anténatal précoce et fiable. La surveillance d’une grossesse suivante se fera par imagerie, en sachant que le diagnostic d’une récidive sera tardif et incertain, car le cervelet se développe tardivement au cours de la grossesse. De plus, certaines de ces pathologies sont évolutives et sans doute non accessibles à un diagnostic anténatal morphologique. Les pathologies associées Les malformations cérébelleuses peuvent s’accompagner d’autres atteintes : neurologiques extracérébelleuses ou extraneurologiques (malformations viscérales, anomalies des extrémités, dysmorphie faciale). Les associations sont des éléments utiles dans la démarche diagnostique car elles peuvent orienter vers un diagnostic syndromique précis. Des aspects cliniques très variables La symptomatologie clinique n’évoque pas toujours d’emblée une atteinte cérébelleuse. Hypotonie Ce symptôme est très souvent au premier plan dans les 2 premières années. Cette hypotonie est présente dès la naissance. Elle se traduit par une mauvaise freination lors de la mobilisation passive des membres. Les réflexes ostéo-tendineux sont difficiles à retrouver, avec fréquemment des réflexes rotuliens pendulaires : le mouvement de balancement de la jambe induit par le réflexe se poursuit exagérément. Cette hypotonie a pour conséquence un retard à la tenue de tête, la station assise, la marche. Ce tableau clinique peut faire errer le diagnostic vers une pathologie neuromusculaire. Troubles oculomoteurs Ces enfants peuvent présenter une apraxie oculomotrice qui est une anomalie de la poursuite oculaire : lors de la fixation d’un objet, le déplacement des yeux s’accompagne d’un mouvement brusque de la tête avec déplacement controlatéral des globes oculaires. Ce signe peut ne pas être facile à observer : on constate parfois uniquement des mouvements brusques de la tête. Dans d’autre cas, on aura l’impression d’un bébé qui ne suit pas bien du regard ou semblant ne pas voir, et le diagnostic de malvoyance est parfois initialement évoqué. Le diagnostic de malvoyance est parfois initialement évoqué. Ataxie cérébelleuse avec ou sans retard intellectuel Parfois, et notamment chez l’enfant plus grand, la classique ataxie cérébelleuse peut être présente. • L’ataxie statique se traduit par des oscillations de la tête chez les plus jeunes, puis du tronc, puis par une instabilité à la station debout puis à la marche, avec élargissement du polygone de sustentation. Cette ataxie est parfois minime, seulement décelable au demi-tour ou à la marche sur une ligne. • L’ataxie cinétique s’observe lors de l’exécution du mouvement : amplitude exagérée du mouvement, mouvements alternatifs rapides difficiles (adiadococinésie mise en évidence par l’épreuve des marionnettes qui est réalisée avec difficulté et lenteur), tremblement intentionnel, écriture lente, tremblée. Cette ataxie s’associe souvent à une atteinte des fonctions supérieures de sévérité très variable. Encéphalopathie sévère Dans certaines étiologies, on observe un tableau d’encéphalopathie sévère : ces enfants ont des acquisitions très limitées. Ils peuvent présenter des signes neurologiques associés : syndrome pyramidal, extrapyramidal, épilepsie, microcéphalie, atteinte de la corne antérieure de la moelle. Cas découverts à l’IRM La malformation est parfois une découverte de l’imagerie cérébrale demandée chez un enfant présentant un retard mental apparemment isolé. Dans les malformations cérébelleuses, le tableau clinique est parfois trompeur : grande hypotonie ou anomalie du regard orientant vers une maladie neuromusculaire ou une malvoyance. Des aspects neuroradiologiques au diagnostic Le diagnostic positif des malformations cérébelleuses a été transformé par l’utilisation de l’IRM cérébrale, aussi bien en postnatal qu’en prénatal devant une anomalie de l’échographie. Classification La classification la plus utilisée repose sur les données radiologiques (voir classification de Ramaekers, figure 1). Figure 1. Classification des anomalies congénitales du cervelet (selon Ramaekers et coll.). Outre le caractère uni- ou bilatéral et l’atteinte exclusive du vermis ou du vermis et des hémisphères cérébelleux, la nature de l’anomalie est parfois difficile à définir avec précision : agénésie du vermis (absence partielle ou totale du vermis), hypoplasie (vermis et/ou hémisphères trop petits mais entiers), atrophie (atteinte secondaire d’un cervelet qui s’était bien développé), atteinte associée ou non du tronc cérébral. Les anomalies unilatérales Elles sont le plus souvent sporadiques, acquises, d’origine vasculaire, sans cause génétique ni métabolique identifiée. Une anomalie chromosomique et des malformations associées pouvant faire suspecter une cause génétique doivent être recherchées systématiquement. Les anomalies prédominant sur le vermis Le syndrome de Dandy- Walker Ce n’est pas une étiologie. La définition est anatomique : hypoplasie du vermis cérébelleux, dilatation kystique de la fosse postérieure communicant avec le 4e ventricule et surélévation de la tente du cervelet (figure 2). Figure 2. Syndrome de Dandy- Walker. Il s’agit d’un groupe hétérogène qui inclut des formes isolées, parfois de bon pronostic, mais aussi des anomalies chromosomiques et des syndromes génétiques. Le syndrome de Joubert Cliniquement, il comporte la triade : atteinte neurologique (hypotonie puis ataxie-retard mental), troubles respiratoires (accès de tachypnée/pauses respiratoires), mouvements oculaires anormaux nystagmiformes. Il peut exister des signes associés : rétinopathie, colobome, hexadactylie, reins kystiques, néphronoptise, fibrose hépatique..., qu’il faut rechercher. L’IRM retrouve une hypoplasie du vermis et « le signe de la molaire » lié à l’hypertrophie et l’horizontalisation des pédoncules cérébelleux supérieurs (figure 3). Il s’agit d’une affection autosomique récessive avec un risque de récurrence de 25 %. Il existe au moins 4 gènes impliqués dont 2 sont identifiés (AHI1 en 6q23, NPHN1 en 2q13) et 2 localisés (9q34.3 et 11p11-q12). Autres étiologies Les autres causes d’hypoplasies du vermis sont les anomalies chromosomiques, les maladies métaboliques, certains syndromes génétiques (orofaciodigital, Smith-Lemli-Opitz, CHARGE, Cornelia de Lange...), les causes environnementales (alcool, CMV, rubéole). Les atteintes des hémisphères et du vermis cérébelleux On regroupe habituellement sous le terme d’hypoplasies cérébelleuses, les anomalies cérébelleuses se traduisant par un cervelet (vermis et hémisphères) insuffisamment développé. Leurs principales étiologies connues sont les causes environnementales ou maternelles (CMV, phénytoïne, radiations ionisantes), les anomalies chromosomiques (trisomies 18 et 21, monosomie 5p et autres), certaines maladies métaboliques (déficit en adénylosuccinase, anomalies de la chaîne respiratoire mitochondriale, les anomalies de la glycosylation des protéines (CDG syndrome), aciduries organiques, des hypo-plasies cérébelleuses familiales (récessive autosomique ou liée à l’X : gène de l’oligophrénine) et des syndromes génétiques. Figure 3. Syndrome de Joubert avec signe de la molaire et hypoplasie du vermis. Les hypoplasies ponto-cérébelleuses (atrophies olivo-pontocérébelleuses) L’hyperplaie cérébelleuse est ici associée à une atteinte de la protubérance (figure 4). Figure 4. Hypoplasie pontocérébelleuse : atteinte du vermis, des hémisphères et du tronc cérébral. On retrouve ici les anomalies mitochondriales, le CDG syndrome (congenital disorders of glycosylation) et des syndromes musculo- oculo-cérébraux (Walker- Warburg, Fukuyama ou Muscle- Eye-Brain syndrom), ainsi que plusieurs entités d’origine génétique, autosomiques récessives que l’on distingue à partir de signes cliniques spécifiques : atteinte de la corne antérieure dans le type I, micro- céphalie progressive et atteinte extrapyramidale dans le type II. Le diagnostic positif des malformations cérébelleuses repose sur l’IRM cérébrale, qui corrélée à la clinique va orienter le diagnostic étiologique. Bilan étiologique Il doit comporter au minimum : - arbre généalogique ; - examen clinique complet (dysmorphie, malformations, croissance) ; - caryotype ; - bilan métabolique : chromatographie des acides aminés (CAA), chromatographie des acides organiques (CAO), lactates, CDG, Saicar-aicar) ; - bilan extraneurologique orienté par l’examen (échographie rénale, électrorétinogramme ; ...) - étude génétique spécifique lorsqu’elle est possible. Un bilan étiologique approfondi incluant anamnèse, clinique et examens spécialisés est indispensable pour tenter de préciser le pronostic et le risque génétique. Conclusion Il faut savoir évoquer la possibilité d’une malformation cérébelleuse devant un tableau clinique qui n’est pas toujours un syndrome cérébelleux typique (hypotonie, encéphalopathie, troubles oculomoteurs). Il est essentiel de s’attacher à identifier une cause précise qui permettra de préciser le pronostic et le conseil génétique, puisqu’il existe pour bon nombre de ces affections un risque de récidive lors d’une nouvelle grossesse.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :