Publié le 14 déc 2010Lecture 9 min
Paralysies focales
D. ARMENGAUD, CHI de Poissy
Voici trois cas cliniques d’enfants ou d’adolescents, chez qui survient de façon soudaine et inexpliquée un déficit moteur apparemment peu grave. Ces troubles neurologiques pourraient bien avoir une origine commune…
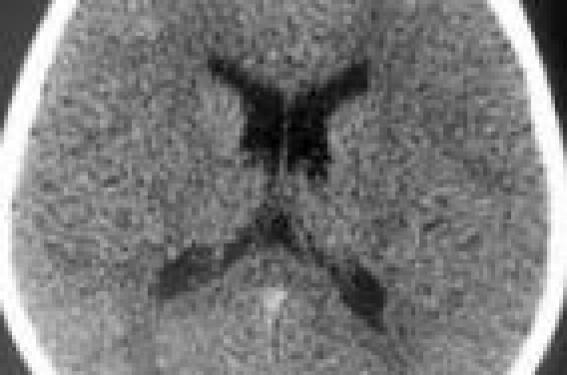
Histoire clinique • Arnaud, 7 ans, est amené en consultation, adressé par son médecin traitant, en raison d’une diminution de la force musculaire du membre supérieure droit constatée par les parents devant des difficultés à relever la main. Quelques jours auparavant, au retour des vacances, les parents notent cette anomalie, qu’il compense spontanément à l’aide de la main gauche. Il n’y a pas de notion de traumatisme, ni d’histoire infectieuse. Figure 1. Arnaud. TDM sans injection : normal. Arnaud ne se plaint de rien, sinon de cette gène dans les gestes de la vie courante. Il ne présente pas d’autres déficit moteur, ni dysesthésie, ni paresthésie, ni céphalées, ni troubles visuels. Un scanner réalisé en urgence en ville est normal (figure 1). Figure 2. Maëlle. IRM cérébrale normale. L’examen clinique retrouve une paralysie flasque, distale avec impossibilité à relever la main ou les doigts, mais leur flexion est possible. Les réflexes cubitaux et radiaux sont absents à droite, mais présents à gauche. Il n’y a pas de troubles de la sensibilité superficielle et profonde. Le reste de l’examen neurologique, notamment les paires crâniennes, et l’examen somatique sont normaux. • Pascale, 12 ans, est amenée par ses parents en raison de la survenue de trois épisodes d’entorse de la cheville gauche au cours des 15 jours précédents, avec impression de difficulté à relever le pied à l’origine de ces épisodes. Une chute de vélo 15 jours auparavant est survenue avec une plaie du genou droit. L’état général est excellent, il n’y a pas de fièvre, ni de syndrome infectieux, et l’examen somatique est normal. L’examen neurologique retrouve un déficit modéré des releveurs du pied à gauche (contre résistance), sans aréflexie, ni signes pyramidaux (Babinski et Rossolimo absents), ni troubles de la sensibilité profonde ou superficielle. Figure 3. Maëlle. IRM médullaire normale. La marche se fait avec un steppage à gauche ; elle est possible mais difficile sur les talons, et impossible sur la pointe à gauche (hypotonie). Il n’y a pas de diminution de la force musculaire au niveau du membre inférieur droit, ni dans les autres territoires. Le reste de l’examen neurologique est normal. Pascale ne se plaint d’aucuns autres troubles (pas de céphalées, paresthésies, dysesthésies, troubles sphinctériens). • Maëlle, 18 ans… moins 1 semaine, consulte aux urgences en raison de paresthésies de l’avant-bras droit constatées le matin même au réveil et s’accompagnant d’une maladresse de la main droite. Elle est apyrétique, en excellent état général avec un examen somatique sans particularité. Cette jeune fille, migraineuse connue, se plaint de céphalées bitemporales pulsatiles, plus fréquentes ces dernières semaines, et soulagées par le paracétamol. L’examen retrouve une diminution de la force musculaire distale du membre supérieur droit avec déficit des releveurs du poignet et des interosseux, alors que la force musculaire au niveau du bras et de l’épaule est normale. Il existe une abolition des réflexes radial et bicipital, seulement à droite. Maëlle se plaint de fourmillement, principalement au niveau du pouce, irradiant vers l’avantbras, sans altération de la sensibilité profonde ou superficielle. Une surveillance au domicile est proposée avec consigne de revenir dans les 48 heures. En fait, elle revient le lendemain en raison d’une aggravation du déficit moteur et de paresthésies de la joue droite et de la face externe de la cuisse droite. Une hospitalisation est décidée pour pratiquer une PL (34 GR, 1 GB, glycorrachie à 3,9 mmol/l, protéinorachie à 0,22 g/l), un bilan biologique qui est normal, une sérologie de Lyme qui reviendra négative et une IRM cérébrale et médullaire, qui s’avèrera parfaitement normale (figures 2 et 3). Ces trois histoires « singulières » sont pourtant en rapport avec une même pathologie. À quoi pensez-vous ? Hypothèses diagnostiques Le point commun de ces trois histoires est la survenue d’une atteinte motrice prédominante, périphérique, touchant la partie plutôt distale d’un membre, supérieur ou inférieur, avec peu ou pas de troubles sensitifs, ou en tout cas sans phénomène douloureux, et sans facteur déclenchant infectieux ou traumatique identifiable. Ces parésies périphériques, peuvent faire évoquer un certain nombre de « radiculonévrite » en l’absence de tout signe évoquant une pathologie du système nerveux central : • Un syndrome de Guillain Barré, qui peut être asymétrique au début, mais rapidement progressif, touchant préférentiellement les membres inférieurs, mais qui s’accompagne de signes neurovégétatifs (dysautonomie) tels qu’une instabilité tensionnelle, et parfois, dans un tiers des cas, d’une atteinte des paires crâniennes (oculomotricité, paralysie faciale), ou surtout de dysesthésies (absentes dans ces trois cas). • Une maladie de Lyme dans sa forme neurologique, possible en cas d’antécédents de morsure de tique oubliée car ancienne, ou passée inaperçue. Mais il n’y a pas de paralysie faciale, ni syndrome méningé ni méningite lymphocytaire (absente en tout cas dans le cas n°3 où la PL a été pratiquée), signes habituellement évocateurs du diagnostic. • Une neuropathie sensitivomotrice, génétique, qui se révèle par une atteinte distale des releveurs, mais dont le caractère progressif est à l’origine de déformations à type de scoliose ou de pieds creux lors du diagnostic, ici absents. Dans ce cadre, c’est la neuropathie de Charcot-Marie- Tooth de type I qui est la plus fréquemment retrouvée dans l’enfance, et dont le diagnostic nécessite EMG et biologie moléculaire, ce d’autant que sa survenue est le plus souvent sporadique. • Une atteinte du plexus brachial (névralgie amyotrophique de l’épaule, plus tardive [adulte jeune]), mais qui reste « localisée ». Un syndrome dysmorphique (épicanthus, syndactylie, fente labiale) est fréquemment présent, ou encore un syndrome de Parsonage et Turner, se manifestant par des douleurs scapulaires à recrudescence nocturne, ou aggravées par le mouvement, avec diminution secondaire de la force musculaire lorsque les douleurs s’atténuent. Reprise de l’interrogatoire C’est la reprise de l’interrogatoire qui va permettre d’évoquer le diagnostic commun de ces trois histoires. • Pour Arnaud, l’apparition de cette parésie est associée à un retour de vacances en voiture la veille, où on apprend qu’il a dormi profondément et longuement, l’épaule droite contre la portière. Au cours de la discussion, le père signale que luimême garde, depuis une intervention chirurgicale sur le pied gauche, une parésie de la main droite ne régressant que très lentement… • Pour Pascale, il est retrouvé une habitude de se tenir assise les jambes croisées pendant de longues heures devant l’ordinateur. Le père signalera qu’il a présenté, il y a plusieurs années, une compression traumatique prolongée de l’avant-bras, suivie d’un déficit d’extension de la main ayant cédé après rééducation kinésithérapeutique. • Pour Maëlle, cette « hémiparésie » est survenue au décours d’une soirée entre amis avec consommation excessive d’alcool et de cannabis, suivie d’un sommeil lourd sur un simple tapis de sol, dans une position de repos sur le côté droit… Le diagnostic à évoquer est celui d’une neuropathie héréditaire avec hypersensibilité à la pression (ou paralysie tomaculaire). Ce diagnostic peut être évoqué devant l’atteinte d’un tronc nerveux favorisée par le maintien anormalement prolongé d’une position comprimant le nerf dans un défilé anatomique, et de ce fait apparaissant souvent au réveil. Cette paralysie est isolée, indolente, de constitution rapide avec hypo-esthésie plus ou moins étendue, et aréflexie transitoire. Les nerfs concernés sont : • le nerf huméral : position latérale pendant le sommeil, bras ballant hors du lit ; • le nerf radial : compression lors du maintien du bras entre dos et dossier (syndrome des amoureux des bancs publics), ou lors du port prolongé d’un nourrisson sur l’avant-bras (allaitement) ; • le nerf cubital : après appui sur le coude prolongé ; • le nerf sciatique poplité externe comprimé sur le col du péroné (position jambes croisées, position assise en tailleur). L’épisode de paralysie évolue habituellement vers la régression, en quelques heures, jours… ou mois, mais la tendance est à la récidive, dans le même territoire ou non, ce qui en fait un élément diagnostique majeur. La persistance peut être à l’origine d’une amyotrophie. Le diagnostic est facilité par la notion d’épisodes de paralysie tronculaire récidivants chez les membres d’une même famille, comme c’était le cas dans deux des trois familles. Commentaire • La neuropathie héréditaire avec sensibilité à la pression, qui a une prévalence de l’ordre de 1/10 000, se manifeste habituellement avant 20 ans par la survenue d’épisodes récurrents concernant un territoire bien défini, mais variable, d’un tronc nerveux avec : – parésie non douloureuse se révélant par une diminution de la force musculaire d’intensité variable, un engourdissement ; – une atteinte des membres, le plus souvent unilatérale, sans atteinte du tronc ou des paires crâniennes ; – des paresthésies, qui sont souvent associées, car il s’agit d’une atteinte sensitivo-motrice ; mais il n’y a jamais de phénomènes douloureux, ce qui en fait un élément d’orientation important. L’épisode peut survenir après un traumatisme, pouvant être minime et passer inaperçu, ou après une compression prolongée. La régression spontanée des troubles en est un des éléments caractéristiques, mais il peut arriver aussi que la répétition conduisent à une amyotrophie, ou entraîne une parésie définitive. • La transmission, sur le mode autosomique dominant, avec forte pénétrance, fait que l’on retrouve très souvent des antécédents de ce type (notamment une paralysie post-opératoire) à l’interrogatoire, ce qui permet le diagnostic sans recourir à de quelconques explorations, nécessaires en cas de forme sporadique. En absence d’antécédents, le diagnostic peut être approché ou affirmé par : – l’EMG, qui montre des signes de dénervation avec allongement des temps de latence motrices distales (> 3,8 ms) et une réduction des potentiels d’action moteurs et sensitifs des nerfs atteints ou au niveau des zones de compression « habituelles » (canal carpien) ; – plus rarement, par une biopsie nerveuse, montrant des zones d’épaississement de la gaine de myéline « en saucisse » à l’origine de la dénomination de paralysie tomaculaire. • Il n’y a pas de traitement autre que préventif, consistant à limiter les durées de compression nerveuse chez les sujets identifiés comme porteur de cette anomalie. • Dans les trois cas présentés, la régression spontanée des troubles moteurs a été observée dans un délai de moins de 3 mois.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :